
Medical device is an instrument, apparatus or machine intended to for the use of diagnosis, treatment or prevention of any disorder. They are divided broadly into three categories depending into their risk levels. Australia serves as a great market for the medical device, and it is estimated to reach USD 6.34bn in 2023 in Australia with the market’s largest contributor being Cardiology Devices with a projected market volume of USD 0.92bn in 2023.
Revenue is expected to show an annual growth rate (CAGR) of 6.08% between 2023-2028, resulting in a market volume of USD 8.52 billion by 2028.
Hence, Australian market serves as a great scope for the development of medical devices for all the local as well as foreign manufacturers.
Medical Device Regulatory Authority in Australia:
Medical devices in Australia shall be approved by the Australian regulatory authority, Therapeutic Goods Administration (TGA). It is the part of Australian government responsible for the safety, efficacy and quality of the therapeutic products.
Any product that intends to be sold in the Australian market needs to be registered with the TGA in the ARTG(Australian Register of Therapeutic Goods) and it is only upon the approval of the TGA a pharmaceutical product can be sold, imported or exported in Australia.
Classification Of Medical Devices In Australia:
The medical devices are classified into four main categories by the Australian classification. They are as follows:
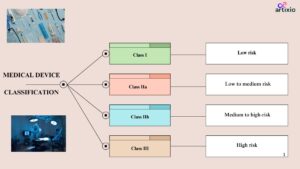
Class I Devices: Low risk devices
- These devices pose low risk and are safe to use as compared to other devices.
- Examples of these devices are bandages, surgical gloves, masks, dental instruments, etc.
- These devices should be labelled with proper name, address of the Australian sponsor/ manufacturer, warnings, precautions, instructions, etc.
Class IIa Devices: Low to Moderate Risk Devices
- These devices pose low to moderate risk level.
- Examples of these devices are surgical instruments, contact lenses, etc.
Class IIb: Medium to High-Risk Devices:
- These devices pose higher risk than the class IIa devices.
- These devices are used in critical body areas for a prolonged duration.
- Examples of this category are implantable devices, in-vitro devices, etc.
Class III Devices: High Risk Devices:
- These are the devices with highest risk associated with them.
- These devices require frequent monitoring and patient assistance.
- These devices include life saving devices such as pacemaker, implantable, etc.
Labelling Requirements For High Risk Devices:
- All the devices should follow the Global Medical Device Nomenclature (GMDN) guidelines as per the Australian guidelines.
- It should consist of detail comprehensive label.
- Additionally, to the class IIa, IIb and III devices’ requirements, class III devices should also include indications for use, contraindications, side effects, storage conditions, etc.
- For devices used in critical body areas, the devices should mention on its label the material used, sterilization and special warnings, etc.
TGA Clinical Trial Pre-requisites For medical Devices:
- Some of the key points to consider before the initiation of the clinical trials are as follows:
- Clinical trials are required for higher risk devices or devices that are novel, and no clinical data is present to support the device.
- For low-risk devices that has clinical data for its support do not need to conduct the clinical trials.
- Researchers must obtain an Investigational Device Exemption (IDE) from the TGA for devices which are not yet approved but the sponsor wants to initiate the trials.
- This IDE should include all the detailed information about the device, its intended use, safety, efficacy and quality parameters.
The IDE application after reviewing by the TGA provides for approval after which the clinical trial can be initiated.
The clinical trials should be conducted with compliance with good manufacturing practise and undertaking informed consent with all the participants.
Medical Product Testing Requirements by TGA
- Medical devices in Australia may need to comply with the regulatory requirements set by the TGA which specify certain testing standards and procedures for medical devices based on their risk classification and intended use. This may include testing for biological safety, electromagnetic compatibility, performance evaluation, and sterilization validation depending on the device type.
- Product testing is typically performed by accredited testing laboratories that have the necessary expertise, equipment, and facilities to conduct the required tests. These laboratories should meet specific accreditation criteria to ensure the reliability and accuracy of their testing results.
Conformity Assessment Certificate:
- The CAB issues a Conformity Assessment Certificate to the manufacturer upon successful completion of the assessment.
- Certification Bodies: For higher-risk devices, the TGA may require the involvement of a third-party Certification Body (CB) in the conformity assessment process.
- Class I devices are exempted from the TGA assessment and do not require conformity assessment.
Medical Device Registration Process In Australia
Local Representative:
A local representative is a local individual/organization who is physically present in Australia and is responsible for all the regulatory procedures on behalf of the foreign manufacturer.
Technical Documentation:
Technical documentation that demonstrates the safety, quality, and performance of the medical device. The documentation includes.
- Medical Device Application
- Technical Files such as device description, intended purpose, design and manufacturing details, labeling, risk assessment, and clinical evidence (if required)
- Australian Declaration of Conformity
- Manufacturer’s Evidence (CE Certificate)
- Appointment of Australian Sponsor
Pre-Market Evaluation
For higher-risk device classes such as Class IIb and III, a pre-market evaluation may be required. Here the various technical documents are evaluated for its compliance with the regulatory requirements.
Submission Of Application:
For attaining the market authorization an application is submitted to the TGA, where the application form is required to be filled on the online portal of ARTG, and the consent documents should be attached along with it.
Australia TGA Medical Devices Registration Review process and Approval:
The TGA reviews and authenticate all the data and document during the review and approval process. If the TGA determines that the device meets the necessary requirements, it will be granted inclusion on the ARTG, which allows the device to be legally supplied and marketed in Australia.
Australia TGA Medical Devices Regulations Registration Timelines: For Class I & II a, typically about 4 weeks, and for Class IIb 6 weeks is required. Class III & Active Implantable Medical Device needs about 6 months.
Annual Charges Applicable:
This is the yearly fee associated with the registration of medical device in the ARTG.
The table below provides the summary for the annual charges applicable for the medical devices:
| Device Class | Annual charges |
| Class III & Active Implantable Medical Device (AIMD) | $1,394.00 |
| Class III | $1,394.00 |
| Class IIa & b | $1,095.00 |
| Class I | $749.00 |
| Other Class I devices | $103.00 |
The US/EU approval is beneficial for gaining Market authorization in Australia, the TGA recognizes the international regulations from the established authorities such as FDA and EMA. TGA accepts conformity assessment reports, clinical data, and risk assessment documents generated during the EU/US approval.
Import and Distribution Requirements:
- The Importers/Distributers should have a valid Medical Device Establishment License (MDEL). The MDEL demonstrates that the establishment has the necessary systems and processes in place to handle and distribute medical devices in accordance with regulatory requirements.
- Before importing or distributing a medical device, it must be included on the ARTG, which is a database of all therapeutic goods authorized for supply in Australia.
- Import medical devices must comply with applicable importation and distribution regulations including compliance with relevant Australian Standards, conformity assessment requirements, labeling and advertising regulations, and post-market obligations.
- Good Distribution Practices: Importers and distributors should adhere to appropriate storage, handling, and distribution practices to ensure the integrity and quality of the medical devices during transportation, storage, and throughout the supply chain.
Post-Marketing Surveillance
After the successful approval of the device and its ale in the Australian market, the manufacturer/sponsor should keep on the monitoring the effects of its device in the patients and keep a record of the adverse effects, safety parameters in the form of a post-marketing report which should be submitted to the TGA.
Adverse Event Reporting: Manufacturers and sponsors are required to report adverse events associated with medical devices to the TGA.
Problem Reporting: Manufacturer/ Sponsors should report any type of problem such as device malfunction, defects or any safety issues related to the device to the TGA.
Corrective And Preventive Action: During the post-marketing surveillance, if any safety or quality issue arises the TGA may demand for a corrective and preventive action to the manufacturer/sponsor. These actions may include issuing field safety notices, conducting device recalls or corrections to mitigate risks associated with the device.
Conclusion:
Thus, we can clearly understand now, that bringing a medical device into market is a task full of struggle. The manufacturer needs to closely comply with all the regulatory requirements of the TGA right from the pre-approval phase till the post-marketing phase. But once regulatory compliance is achieved, Australian market serves as a great scope for developmental success with medical devices.
For more details on the regulatory process, get in touch with Artixio, a globally trusted consultant for regulatory assistance.
FAQs – Australia (TGA) Regulations for Medical Device Registration
Here are answers to some of the Frequently Asked Questions (FAQs) about Australia regulations for Medical Device registration.
What is the regulatory authority responsible for medical device registration in Australia?
The regulatory authority responsible for medical device registration in Australia is the Therapeutic Goods Administration (TGA).
The TGA is a division of the Australian Government Department of Health and Ageing, and is responsible for ensuring the safety, quality and efficacy of therapeutic goods (including medical devices) in Australia.
What are the steps involved in the registration process of medical device in Australia as per TGA regulations?
- Determine the risk classification of your device. Medical devices are classified into six risk classes: Class I, Class IIa, Class IIb, Class III, Class IV, and Class V. The risk classification of your device will determine the evaluation route and the documentation requirements for registration.
- Choose the evaluation route. There are two evaluation routes for medical device registration in Australia: Standard Evaluation Route and Simplified Evaluation Route. The evaluation route that you choose will depend on the risk classification of your device, the availability of clinical data, and the prior approvals received from overseas regulatory agencies.
- Prepare the registration dossier. The registration dossier is a collection of documents that demonstrate that your device meets the safety, quality, and efficacy requirements of the TGA. The contents of the registration dossier will vary depending on the evaluation route that you choose. The documents required in a medical device registration dossier for TGA in Australia vary depending on the risk classification of the device. However, some of the common documents that are required include:a) Summary of Safety and Efficacy (SoSE): This document provides an overview of the safety and efficacy of the device. It should include information about the device’s intended use, the results of any clinical trials, and the safety data.
b) Technical File (TF): This document provides detailed information about the device, its design, manufacturing, and testing. It should include information about the materials used in the device, the manufacturing process, and the test methods used to assess the device’s safety and efficacy.
c) Labeling and Instructions for Use (IFU): This document provides information about how to use the device safely and effectively. It should include information about the device’s intended use, the risks associated with the device, and the instructions for cleaning and maintenance.
d) Quality Management System (QMS): This document describes the quality management system (QMS) that is used to manufacture the device. It should include information about the QMS’s objectives, procedures, and controls.e) Clinical Evaluation Report (CER): This document is required for Class III and Class IV devices. It provides an assessment of the clinical data that is available for the device.
f) Additional documents: Depending on the risk classification of the device, other documents may be required, such as a risk management file (RMF) or a post-market surveillance plan (PMSP).
- Submit the registration application. The registration application can be submitted online through the TGA’s ARTG (Australian Register of Therapeutic Goods) website. The application fee will vary depending on the risk classification of your device.
- Review and approval of the application. The TGA will review your application and make a decision on whether to register your device. The review process can take several months.
- Issuance of the registration certificate. If your device is registered, you will be issued a registration certificate. You will need to display this certificate on your device and on all marketing materials for your device.
Are there any specific labeling or packaging requirements for medical devices in Australia as per TGA regulations?
Yes, there are specific labeling and packaging requirements for medical devices in Australia as per TGA regulations. These requirements are designed to ensure that medical devices are properly labeled and packaged so that they can be used safely and effectively.
The labeling requirements for medical devices in Australia are set out in the Therapeutic Goods (Labelling) Regulations 2008. The labeling must include the following information:
- The name and address of the manufacturer, packer, or distributor
- The trade or brand name of the device
- The unique device identifier (UDI)
- The intended use of the device
- The instructions for use
- Any warnings or precautions
- The expiry date (if applicable)
- The batch or lot number
The packaging requirements for medical devices in Australia are set out in the Therapeutic Goods (Packaging) Regulations 2008. The packaging must be designed to protect the device from damage and to prevent it from being used incorrectly. The packaging must also include the following information:
- The name and address of the manufacturer, packer, or distributor
- The trade or brand name of the device
- The unique device identifier (UDI)
- The intended use of the device
- The instructions for use
- Any warnings or precautions
- The expiry date (if applicable)
- The batch or lot number
In addition to the labeling and packaging requirements, medical devices in Australia must also comply with the Therapeutic Goods (Quality Management) Regulations 2002. These regulations require manufacturers to have a quality management system (QMS) in place to ensure that the devices are manufactured and tested to a high standard.
Are clinical trials or testing necessary for registration of medical devices in Australia? If so, what are the TGA requirements?
Clinical trials or testing are not always necessary for registration of medical devices in Australia. However, the TGA may require clinical trials or testing depending on the risk classification of the device and the intended use of the device.
- For Class I medical devices, clinical trials or testing are not usually required. However, the TGA may require clinical trials or testing if the device is intended for a new use or if there are concerns about the safety or efficacy of the device.
- For Class IIa medical devices, clinical trials or testing may be required, depending on the intended use of the device. For example, clinical trials or testing may be required if the device is intended for a new use or if there are concerns about the safety or efficacy of the device.
- For Class IIb medical devices, clinical trials or testing are usually required. The TGA will need to be satisfied that the device is safe and effective for its intended use before it will be registered.
- For Class III and Class IV medical devices, clinical trials or testing are always required. The TGA will need to be satisfied that the device is safe and effective for its intended use before it will be registered.
Some examples of Clinical Trial conducted in Australia for Class III/IV devices:
The PRECISE Trial: This trial was conducted to assess the safety and efficacy of the PRECISE stent for the treatment of coronary artery disease. The trial involved 1,200 patients who were randomly assigned to receive either the PRECISE stent or a standard stent. The results of the trial showed that the PRECISE stent was safe and effective in reducing the risk of heart attack and death.
The ESCAPE Trial: This trial was conducted to assess the safety and efficacy of the ESCAPE pacemaker for the treatment of heart failure. The trial involved 2,010 patients who were randomly assigned to receive either the ESCAPE pacemaker or a standard pacemaker. The results of the trial showed that the ESCAPE pacemaker was safe and effective in reducing the risk of death and hospitalization for heart failure.
The MIRACLE-ICD Trial: This trial was conducted to assess the safety and efficacy of the MIRACLE-ICD implantable cardioverter defibrillator (ICD) for the treatment of sudden cardiac death. The trial involved 4,082 patients who were randomly assigned to receive either the MIRACLE-ICD or a standard ICD. The results of the trial showed that the MIRACLE-ICD was safe and effective in reducing the risk of sudden cardiac death.
What are the fees associated with the medical devices registration process in Australia as per TGA regulations?
Registration application fee: The registration application fee for medical devices in Australia varies depending on the risk classification of the device.
- Class I: AUD 2,150
- Class IIa: AUD 3,200
- Class IIb: AUD 4,250
- Class III: AUD 5,300
- Class IV: AUD 6,350
Annual retention fee: The annual retention fee for medical devices in Australia also varies depending on the risk classification of the device.
- Class I: AUD 550
- Class IIa: AUD 700
- Class IIb: AUD 850
- Class III: AUD 1,000
- Class IV: AUD 1,150
In addition to the registration application fee and the annual retention fee, there may be other costs associated with the registration process, such as the cost of preparing the registration dossier and the cost of clinical trials or testing.
Is it necessary to have a local authorized representative in Australia for Medical Device registration?
Yes, it is necessary to have a local authorized representative (LAR) in Australia for medical device registration. The LAR is a company or individual who is appointed by the foreign manufacturer to represent them in Australia. The LAR is responsible for submitting the registration application to the TGA, responding to TGA queries, and providing the TGA with updates on the device.
- The LAR must be a company or individual that is registered with the TGA and that has a valid dealer’s licence. The LAR must also have a qualified person responsible for regulatory affairs (QPR) who is familiar with the TGA regulations for medical devices.
- The LAR plays an important role in the medical device registration process in Australia. By having a LAR, the foreign manufacturer can ensure that their device is registered in Australia and that they are compliant with the TGA regulations.
Here are some of the benefits of having a local authorized representative in Australia for medical device registration:
- The LAR can help you navigate the registration process and ensure that your application is compliant with TGA requirements.
- The LAR can represent you in dealings with the TGA, which can save you time and effort.
- The LAR can provide you with local market insights and help you build relationships with key stakeholders in Australia.
Artiixo can act as your LAR in Australia. Please email to info@artixio.com for queries related to services.
Are there any post-registration obligations or reporting requirements for medical devices as per TGA regulations in Australia?
Yes, there are a number of post-registration obligations or reporting requirements for medical devices as per TGA regulations in Australia. These obligations and requirements are designed to ensure that medical devices remain safe and effective throughout their life cycle.
Some of the key post-registration obligations and reporting requirements include:
- Change notification: If you make any changes to your device, you must notify the TGA within 30 days of the change. This includes changes to the device’s design, manufacturing process, or labeling.
- Adverse event reporting: You must report any adverse events (AEs) associated with your device to the TGA within 15 days of becoming aware of the AE. An AE is any undesirable or unexpected event that occurs during the use of a medical device, and that could potentially harm the patient.
- Field safety corrective action (FSCA): If you become aware of a safety issue with your device, you must take steps to correct the issue. This may involve issuing a recall of the device, or implementing other corrective actions.
- Product evaluation: You must periodically evaluate your device to ensure that it remains safe and effective. This evaluation should include a review of the device’s design, manufacturing process, and clinical data.
Are there any exemptions or expedited processes available for certain types of medical devices as per TGA regulations in Australia?
Yes, there are a number of exemptions or expedited processes available for certain types of medical devices as per TGA regulations in Australia. These exemptions and expedited processes are designed to facilitate the registration of medical devices that are considered to be low-risk or that have already been approved by other regulatory authorities.
Some of the key exemptions and expedited processes include:
- Exemption for Class I devices: Class I devices are considered to be low-risk devices, and they are exempt from the full registration process. These devices can be registered through a simplified registration process.
- Expedited registration for devices approved by other regulatory authorities: If your device has already been approved by another regulatory authority that is considered to be equivalent to the TGA, you may be eligible for expedited registration. This means that your device can be registered in Australia without the need to submit a full registration dossier.
- Special access routes (SARs): SARs are available for devices that are urgently needed in Australia, but that have not yet been approved by the TGA. SARs allow these devices to be used on patients in Australia under certain conditions.
Are there any unique considerations or requirements for software or digital health applications in Australia as per TGA regulations?
Some of the key considerations or requirements include:
- Risk classification: Software or digital health applications are classified into four risk classes: Class I, Class IIa, Class IIb, and Class III. The risk class of the application will determine the evaluation route and the documentation requirements for registration.
- Cybersecurity: Software or digital health applications must be designed and developed with cybersecurity in mind. This includes ensuring that the application is protected from unauthorized access, use, disclosure, disruption, modification, or destruction.
- Data protection: Software or digital health applications must comply with the Australian Privacy Principles (APPs). This means that the application must be used in a way that protects the privacy of the data that it collects or processes.
- Clinical evaluation: For Class III and Class IV applications, a clinical evaluation report (CER) must be submitted to the TGA. The CER must demonstrate that the application is safe and effective for its intended use.
- Labeling and packaging: Software or digital health applications must be labeled and packaged in a way that is clear, accurate, and informative. The labeling must include information about the application’s intended use, the risks associated with the application, and the instructions for use.
Are there any local standards or additional testing requirements beyond international standards for medical devices in Australia per TGA regulations?
- Australian Standards: The TGA recognizes a number of Australian Standards (AS) for medical devices. These standards provide guidance on the design, manufacture, and testing of medical devices.
- TGA-approved conformity assessment bodies: The TGA has approved a number of conformity assessment bodies (CABs) to assess the safety and efficacy of medical devices. These CABs are required to follow the TGA’s guidelines for conformity assessment.
- Additional testing: In some cases, the TGA may require additional testing of medical devices to ensure that they meet the specific needs of the Australian market. This testing may be conducted by the manufacturer or by an independent laboratory.
Can I use a foreign clinical study or data for medical devices registration purposes in Australia per TGA regulations?
Yes, you can use a foreign clinical study or data for medical devices registration purposes in Australia per TGA regulations. However, there are a number of factors that you need to consider before doing so.
Factors to consider:
- The risk classification of the device: The risk classification of the device will determine the extent to which you can rely on foreign clinical data. For example, you may be able to rely on foreign clinical data for Class I devices, but you may need to conduct additional clinical trials for Class C or D devices.
- The similarity of the population: The population that was studied in the foreign clinical trial should be similar to the population that will be using the device in Australia. If the populations are not similar, you may need to conduct additional clinical trials in Australia.
- The availability of local data: If there is local clinical data available, you should generally use the local data. This is because the local data will be more relevant to the Australian market.
How to use foreign clinical study or data:
If you decide to use a foreign clinical study or data, you will need to submit a justification to the TGA. The justification should include information about the following:
- The risk classification of the device
- The similarity of the population
- The availability of local data
- The reasons why you believe that the foreign clinical data is relevant to Australia
- The TGA will review your justification and make a decision on whether to allow you to use the foreign clinical data.
How can the adverse effects be reported to TGA?
The adverse reactions can be reported by the using the “Incident reporting” system developed by the TGA.
How can a product be checked if it’s registered with TGA or not?
The product can be searched in the Australian Register of Therapeutic Goods (ARTG) on the TGA website to confirm if it’s registered with TGA or no.
What are “listed” and “registered” medicines. Are both the categories different?
Yes, listed and registered medicines are different from each other. Listed medicines includes nutraceuticals, herbal supplements, etc that are medicines with low risk whereas registered medicines include high risk medicines such as anti-inflammatory medicines, antibiotics, etc.
Did you find what you were looking for? Join us to follow more Frequently Asked Questions about global regulations for medical devices.
References:
- https://www.tga.gov.au/
- https://www.health.gov.au/contacts/therapeutic-goods-administration-tga
- https://clinregs.niaid.nih.gov/country/australia



