
The introduction of the EU Medical Device Regulation (MDR) has brought increased emphasis on robust data and comprehensive evaluations for ensuring the safety of medical devices. Under MDR, the subclass for Class I reusable devices (Class Ir), including surgical instruments and endoscopes, are. subjected to stricter scrutiny and enhanced regulatory oversight. Class Ir devices must adhere to the new guidelines or risk exclusion from the market.
The MDR defines a reusable surgical instrument as “instruments intended for surgical use in cutting, drilling, sawing, scratching, scraping, clamping, retracting, clipping or similar procedures, without a connection to an active device and which is intended by the manufacturer to be reused after appropriate procedures such as cleaning, disinfection and sterilization have been carried out.” (Annex VIII, Chapter 1, 2.3).
What has changed from EU MDD –
Unlike the previous European Medical Device Directive (MDD), Class Ir devices now require a detailed technical file to obtain a CE Mark under MDR. This file must demonstrate the safety and efficacy of cleaning, disinfection, and sterilization methods outlined in the device’s Instructions for Use (IFU). It’s no longer a self-designation process; every manufacturer is responsible for ensuring the adequacy and validation of these instructions. Notified bodies will review these technical files, including validations, to ensure MDR compliance based on international standards and industry guidance. Key changes from EU MDD to MDR include –
- New classifications for reusable surgical devices with notified body oversight.
- A broader definition of medical devices, covering non-medical and cosmetic devices not previously regulated.
- Expanded scope of cleaning instructions and validations, including disinfection, sterilization, maintenance, and functional testing.
- No “grandfathering” provision; previously approved devices must be recertified under the new rules
The Article 52(7c) of EU MDR mandates manufacturers of reusable surgical instruments to follow procedures set out in Chapters I and III of Annex IX, or in Part A of Annex XI and the Notified Body involvement is required only regarding cleaning, disinfection, sterilization, maintenance, and functional testing, as well as providing instructions for use. Manufacturers must develop and maintain a Technical File, apply appropriate Conformity Assessment Procedures, issue a Declaration of Conformity, and affix the CE Mark and notified body number.
Regulation Compliance Pathway of Reusable Medical Devices under EU MDR
To achieve compliance, manufacturers of Class Ir devices must identify data gaps and select appropriate validation studies to demonstrate compliance with MDR.


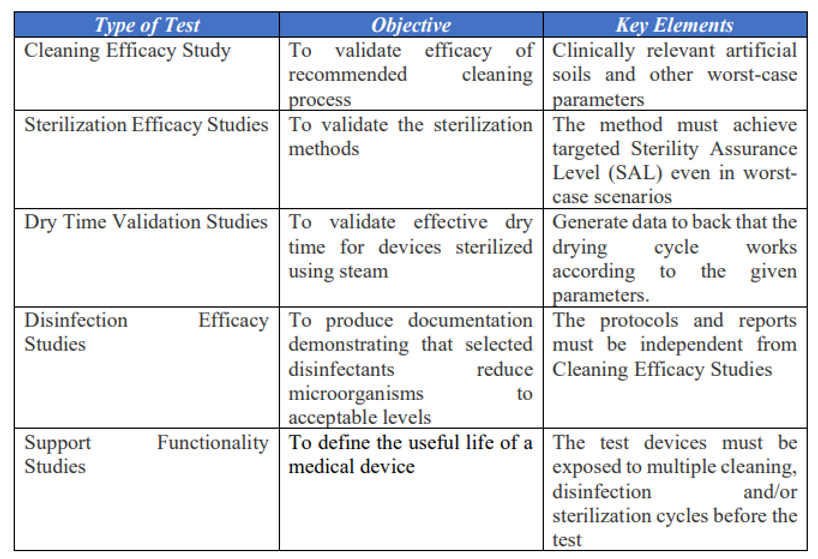
Validation Studies for Reusable Medical Devices In EU
To validate the effectiveness of a process design and ensure that process variables do not impact device acceptance criteria for reusability, a cleaning validation process is necessary. This validation package should confirm that both manual and mechanical cleaning methods allow medical devices to be efficiently reprocessed. For devices cleaned in healthcare facilities, proper cleaning instructions must be provided to users in those facilities.
The primary goal is to create evidence and documentation showing that the reusability processing methods consistently produce intended outcomes. Key validation principles that organizations should incorporate include:
- Robustness: This involves testing how a device performs in worst-case scenarios.
- Repeatability: It assesses measurement variability, ensuring data consistency and process consistency for specified outputs.
- Reproducibility: This examines possible variations in the system and supports data integrity. Multiple effectiveness cycles are used to explain variations in the test system, and it verifies whether the validation study can be replicated with the same results.
Key consideration while conducting studies
Inappropriately bundling a validation study for reusable devices can lead to significant time and financial setbacks. Failure to provide adequate validation can result in delays in reaching the market, increased overall pre-clinical testing costs, and a potential loss of market share. Given the substantial investment of both time and resources required for these studies, it is imperative to conduct them meticulously and comprehensively from the outset.
- It’s essential to be mindful of the following common reasons why validation studies for reusable devices may fail:
- Ensure the selection of clinically relevant test soils, including worst-case validation soil tests.
- Simulate worst-case conditions and incorporate worst-case devices and parameters.
- Implement appropriate test controls.
- Utilize two or more suitable cleaning endpoint markers.
- Include accumulation cycles in the validation process.
- Assess recovery efficiency and address challenges if the recovery effectiveness is too low.
- Conduct validations independently; avoid combining cleaning and disinfection validation in a single test.
- Demonstrate and meticulously document appropriate test soil drying times.
These precautions are essential to ensure the accuracy and effectiveness of your validation studies for reusable devices, helping you avoid costly setbacks and maintain the integrity of product development process.
Reusable devices under EU MDR now need more than a tick-box approach. What was acceptable under MDD often isn’t enough anymore, especially when it comes to reprocessing instructions and validation data. Manufacturers are expected to clearly show how their devices can be cleaned, disinfected, sterilized, and safely reused — and to back this up with proper studies and documentation. This is where many projects slow down or run into avoidable questions from notified bodies.
EU MDR reviews can get difficult fast, especially for reusable devices. Questions often come up around cleaning validation, reprocessing steps, or missing details in the technical file. Sorting these issues early helps avoid delays later. Artixio supports manufacturers with EU regulatory affairs work, focusing on what notified bodies actually look for during review.
Explore more about our EU regulatory affairs consulting services and contact us at info@artixio.com

