
The healthcare landscape is in a perpetual state of transformation, and as a consequence, there is a noticeable surge in the development of innovative combination products engineered to offer ground-breaking therapies and solutions. These combination products present distinctive opportunities and challenges, particularly in the context of regulatory scrutiny, approval processes, and subsequent post-approval compliance requirements.
US FDA RFD and Pre-RFD Submissions for Combination Products in USA
A combination product is defined as a product that encompasses two or more regulated components, which can be drugs, devices, or biologics. These combinations can fall into several categories:
● Drug and Device
● Biological Product and Device
● Drug and Biological Product
● Drug, Device, and Biological Product
These combination products may be introduced to the market with their components either bundled into a single entity or individually packaged but labeled for co-use.
The US FDA functions via specialized divisions, namely CDER, CBER, and CDRH, each responsible for overseeing Drugs, Biologics, and Medical Devices, respectively. However, certain products challenge these distinctions, creating ambiguity and blurring the lines between them.
The US FDA has established the Office of Combination Products (OCP) to handle regulatory considerations related to Combination Products. In instances where ambiguity arises, the FDA has put in place processes to resolve such issues and to determine which FDA center should regulate the product. Manufacturers of combination products can seek clarity by making use of the FDA’s RFD (Request for Designation) or Pre-RFD (Pre-Request for Designation) submissions. These processes play a pivotal role in ascertaining the correct regulatory path and facilitating communication with the OCP.
Manufacturers have the option to approach the OCP to classify their combination product and identify the lead center responsible for pre-market review submissions. The OCP utilizes two distinct processes for this purpose:
● The Pre-Request for Designation (Pre-RFD)
● The Request for Designation (RFD)
Operating under Section 503 (g) (1) of the Federal Food, Drug, and Cosmetic Act (the Act) (21 U.S.C. 353(g)(1)), the OCP assigns review responsibilities based on the ‘primary mode of action’ of the product.”
Key Benefits of the RFD and Pre-RFD Processes
Regulatory Clarity: These processes provide clarity on which agency center will regulate the combination product, ensuring a more streamlined and efficient regulatory pathway.
Early Engagement: Sponsors can engage with the FDA early in the development process to get insights and feedback, helping them make informed decisions and navigate the regulatory landscape more effectively.
Enhanced Communication: By fostering open communication with the FDA, sponsors can address concerns and resolve potential issues early, reducing delays in the development and approval process.
Regulatory Compliance: Determining the correct regulatory pathway ensures that combination products meet the necessary safety and efficacy standards, enhancing patient safety.
Pre-Request for Designation (Pre-RFD)
The Pre-Request for Designation (Pre-RFD) procedure provides manufacturers with the opportunity to obtain informal input from the FDA prior to formally submitting their product registration application. Pre-RFD submissions are not subject to any specific length constraints and do not create binding obligations on the agency or the sponsor. These submissions can be made at any stage of the medical product development process. The US FDA typically takes 60 calendar days to provide feedback on the application.”
Request for Designation (RFD)
The Request for Designation (RFD) process, as detailed in 21 CFR Part 3, provides manufacturers with the opportunity to obtain official input from the FDA prior to initiating any investigational or marketing application for their product. RFD submissions are limited to a maximum of 15 pages. The designation resulting from the RFD is legally binding on both the agency and the sponsor. These submissions can be initiated at any phase during the development of a medical product. The US FDA typically takes 60 calendar days from the time they receive essential information to make a determination on Product Designation. Sponsors are required to furnish an assessment of the product’s classification, a primary mode of action (PMOA) analysis (if the RFD pertains to the assignment of a combination product), and a recommendation concerning Agency Center assignment.
Information to be included in RFD and Pre-RFD Submissions
The fundamental elements to be incorporated in RFD and Pre-RFD applications consist of
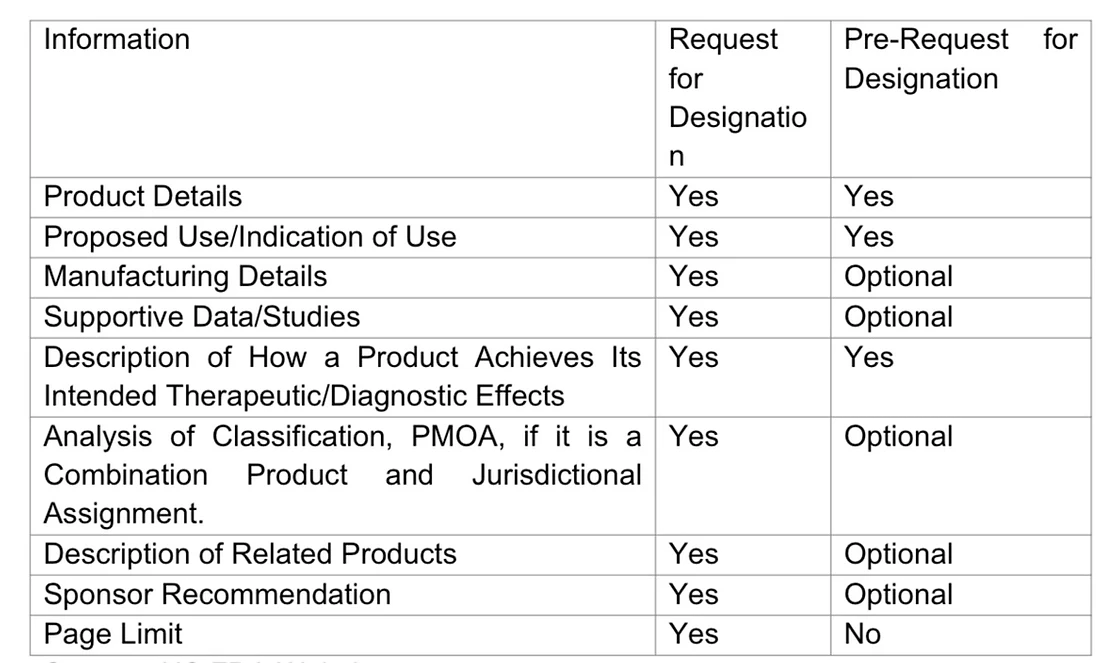
Source : US FDA Website
Combination products hold great promise for improving patient care and advancing healthcare. However, their regulatory complexity can be a challenge for product developers. The Request for Designation (RFD) and Pre-RFD processes established by the US FDA are essential tools to ensure that these innovative products are evaluated and regulated appropriately. Early engagement with the FDA can help sponsors navigate the regulatory landscape more effectively, leading to better outcomes for patients and the healthcare industry as a whole. As the world of combination products continues to evolve, these processes play a vital role in ensuring their safety, efficacy, and market access.
Ready to navigate the world of combination products and ensure your innovation gets the regulatory approval it deserves? Whether you’re a product developer, healthcare professional, or simply curious about the FDA’s RFD and Pre-RFD processes, we’re here to help. Contact Artixio for expert guidance and assistance in bringing your combination product to market. Don’t miss out on the opportunity to make a difference in patient care – reach out to Artixio today!

