
Australia’s medical device market is sizeable—about USD 6.34bn in 2023, with cardiology devices near USD 0.92bn—and is projected to hit USD 8.52bn by 2028 (6.08% CAGR). But growth doesn’t equal easy entry: success starts with understanding Australia medical device regulations and completing medical device registration in Australia.
But tapping into this Australian market isn’t just about innovation—it starts with meeting Australia’s medical device regulations and registration requirements.
Medical Device Regulatory Authority in Australia
Medical devices in Australia are approved by the Australian regulatory authority, Therapeutic Goods Administration (TGA). Any medical device before selling or importing in the Australian market needs to be registered with the TGA in the ARTG(Australian Register of Therapeutic Goods). Only after approval can the device legally enter the Australian market.
Classification Of Medical Devices In Australia
Medical device classification in Australia are classified into four main categories by TGA. They are as follows:
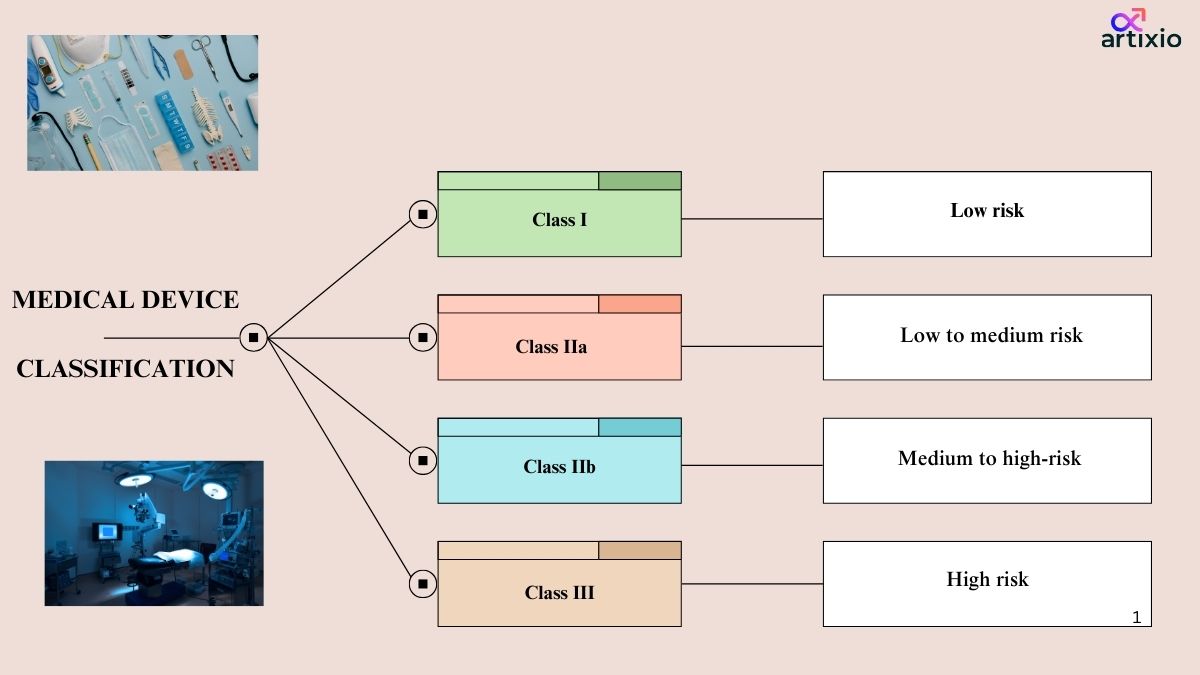
Class I Devices: Low risk devices
- These devices pose low risk and are safe to use as compared to other devices.
- Examples of these devices are bandages, surgical gloves, masks, dental instruments, etc.
- These devices should be labelled with proper name, address of the Australian sponsor/ manufacturer, warnings, precautions, instructions, etc.
Class IIa Devices: Low to Moderate Risk Devices
- These devices pose low to moderate risk level.
- Examples of these devices are surgical instruments, contact lenses, etc.
Class IIb: Medium to High-Risk Devices:
- These devices pose higher risk than the class IIa devices.
- These devices are used in critical body areas for a prolonged duration.
- Examples of this category are implantable devices, in-vitro devices, etc.
Class III Devices: High Risk Devices:
- These are the devices with highest risk associated with them.
- These devices require frequent monitoring and patient assistance.
- These devices include life saving devices such as pacemaker, implantable, etc.
Medical Device Registration Process In Australia
Local Representative:
A local representative is a local individual/organization who is physically present in Australia and is responsible for all the regulatory procedures on behalf of the foreign manufacturer.
Technical Documentation:
Below Documents showing that the medical device is safe, works properly, and meets quality standards.
- Medical Device Application
- Technical Files such as device description, intended purpose, design and manufacturing details, labeling, risk assessment, and clinical evidence (if required)
- Australian Declaration of Conformity
- Manufacturer’s Evidence (CE Certificate)
- Appointment of Australian Sponsor
Pre-Market Evaluation
For higher-risk device classes such as Class IIb and III, a pre-market evaluation may be required. Here the various technical documents are evaluated for its compliance with the regulatory requirements.
Submission Of Application:
For attaining the market authorization an application is submitted to the TGA, where the application form is required to be filled on the online portal of ARTG, and the consent documents should be attached along with it.
Australia TGA Medical Devices Registration Review process and Approval:
The TGA reviews and authenticate all the data and document during the review and approval process. If the TGA determines that the device meets the necessary requirements, it will be granted inclusion on the ARTG, which allows the device to be legally supplied and marketed in Australia.
Australia TGA Medical Devices Regulations Registration Timelines: For Class I & II a, typically about 4 weeks, and for Class IIb 6 weeks is required. Class III & Active Implantable Medical Device needs about 6 months.
Australia Medical Device Labeling Requirements
Here are the TGA Labelling Requirements For High Risk Devices in Australia:
- All the devices should follow the Global Medical Device Nomenclature (GMDN) guidelines as per the Australian guidelines.
- Device should consist of detail comprehensive label.
- Additionally, to the class IIa, IIb and III devices’ requirements, class III devices should also include indications for use, contraindications, side effects, storage conditions, etc.
- For devices used in critical body areas, the devices should mention on its label the material used, sterilization and special warnings, etc.
Annual Charges Applicable:
This is the yearly fee associated with the registration of medical device in the ARTG.
The table below provides the summary for the annual charges applicable for the medical devices:
| Device Class | Annual charges |
| Class III & Active Implantable Medical Device (AIMD) | $1,394.00 |
| Class III | $1,394.00 |
| Class IIa & b | $1,095.00 |
| Class I | $749.00 |
| Other Class I devices | $103.00 |
The US/EU approval is beneficial for gaining Market authorization in Australia, the TGA recognizes the international regulations from the established authorities such as FDA and EMA. TGA accepts conformity assessment reports, clinical data, and risk assessment documents generated during the EU/US approval.
TGA Clinical Trial Requirements For medical Devices:
Some of the key points to consider before the initiation of the clinical trials for a medical device in Australia are as follows:
- Trials are needed for high-risk or new devices when no supporting clinical data is available.
- Low-risk devices with existing supporting data generally don’t require trials.
- Before testing a device that isn’t approved yet, you must get permission from the TGA called an IDE.
- This IDE should show what the device does, how it will be used, and any proof that it’s safe and works properly.
- After reviewing the IDE, the TGA may grant approval, allowing the trial to begin.
- Trials must be run under good practice standards, and all participants must give informed consent.
Medical Product Testing Requirements by TGA
Medical devices in Australia may need to follow TGA rules, which decide what tests are needed based on the device’s risk and how it will be used. Tests are done in qualified labs that have the right equipment and know-how. They make sure the device works safely and as intended.
Conformity Assessment Certificate
- After passing the assessment, the manufacturer gets a Conformity Assessment Certificate from the CAB.
- High-risk devices may need a third-party Certification Body.
- Class I devices don’t need TGA assessment or certification.
Import and Distribution Requirements:
- Importers and distributors need a valid Medical Device Establishment License (MDEL).
All devices must be listed on the ARTG before supply. - Devices should meet regulatory rules, labeling standards, and post-market obligations.
Proper storage and handling must be followed to keep devices safe and effective.
Post-Marketing Surveillance
After a device is approved, the manufacturer or sponsor must track its performance in patients and report any issues to the TGA.
- Adverse Events: Report any side effects or safety concerns.
- Device Problems: Report malfunctions, defects, or other issues.
- Corrective Actions: If problems arise, the TGA may require recalls, safety notices, or fixes to reduce risk.
Conclusion:
Thus, we can clearly understand now, that bringing a medical device into market is a task full of struggle. The manufacturer needs to closely comply with all the regulatory requirements of the TGA right from the pre-approval phase till the post-marketing phase. But once regulatory compliance is achieved, Australian market serves as a great scope for developmental success with medical devices.
For more details on the regulatory process, get in touch with Artixio, a globally trusted consultant for regulatory assistance.
FAQs – Australia (TGA) Regulations for Medical Device Registration
Here are answers to some of the Frequently Asked Questions (FAQs) about Australia regulations for Medical Device registration.
Do You Need a Local Representative For Device Registration in Australia?
If your company is outside Australia, you need someone on the ground—a Local Authorized Representative (LAR). They handle the paperwork with the TGA, answer questions, and keep the authorities updated about your device.
The LAR must be registered and have someone who knows the rules well. Having a local contact also makes it easier to deal with the TGA and understand the Australian market.
Artiixo can act as your LAR in Australia. Please email to info@artixio.com for queries related to services.
Are there any local standards or additional testing requirements beyond international standards for medical devices in Australia per TGA regulations?
- Australian Standards: The TGA recognizes a number of Australian Standards (AS) for medical devices. These standards provide guidance on the design, manufacture, and testing of medical devices.
- TGA-approved conformity assessment bodies: The TGA has approved a number of conformity assessment bodies (CABs) to assess the safety and efficacy of medical devices. These CABs are required to follow the TGA’s guidelines for conformity assessment.
- Additional testing: In some cases, the TGA may require additional testing of medical devices to ensure that they meet the specific needs of the Australian market. This testing may be conducted by the manufacturer or by an independent laboratory.
How can the adverse effects be reported to TGA?
The adverse reactions can be reported by the using the “Incident reporting” system developed by the TGA.
How can a product be checked if it’s registered with TGA or not?
The product can be searched in the Australian Register of Therapeutic Goods (ARTG) on the TGA website to confirm if it’s registered with TGA or no.
What are “listed” and “registered” medicines. Are both the categories different?
Yes, both listed and registered medicines are different. Listed medicines are low-risk products, like herbal supplements or vitamins. They don’t need strict approval before being sold. Registered medicines are higher-risk drugs, such as antibiotics or anti-inflammatory medicines, and they must go through a thorough review before they can be sold.

