Although the manufacturing sector for medical devices in Indonesia is constantly growing, Indonesia is an import-driven market for advanced and specialized medical equipment.
Increased awareness of healthcare issues and an increase in the elderly population aged above 60, have led to greater demand for advanced medical devices and technology in Indonesia.
Medical Devices Regulations and Registration in Indonesia
The regulatory authority responsible for overseeing medical devices in Indonesia is the Indonesian National Agency of Drug and Food Control, known as Badan Pengawas Obat dan Makanan (BPOM). BPOM is an Indonesian government agency under the ‘Ministry of Health’ that plays a crucial role in ensuring the quality of medical devices in the country.
Medical device registration in Indonesia involves multiple regulatory touchpoints. Our team works alongside manufacturers to manage BPOM submissions and local requirements. Contact Artixio today!
Medical Device Classification in Indonesia:
The classification system is similar to the one used by the Global Harmonization Task Force (GHTF) and the Association of Southeast Asian Nations (ASEAN).
Class A: Low-risk medical devices with a low potential to cause harm. Examples include adhesive bandages, tongue depressors, and examination gloves.
Class B: Low to moderate-risk medical devices with a moderate potential to cause harm. Examples include non-invasive diagnostic devices like blood pressure monitors and thermometers.
Class C: Moderate to high-risk medical devices that could pose a significant risk to patients if they malfunction. Examples include surgical instruments, certain types of catheters, and in-vitro diagnostic devices.
Class D: High-risk medical devices that could pose serious health risks if they fail to function as intended. Examples include implantable devices like pacemakers and artificial joints.
Labeling Requirements as per Medical Device Regulations and Registration in Indonesia
Class A Medical Devices:
- Product Name and Model/Catalog Number
- Manufacturer Information (Name and Address)
- Instructions for Use
- Precautions and Warnings, if applicable
- Batch/Serial Number, if applicable
- Expiry Date, if applicable
- Storage and Handling Information, if applicable
- Regulatory Information (Registration number, if applicable)
Class B Medical Devices:
In addition to the requirements for Class A devices, Class B, C, and D medical devices might have more specific and comprehensive labeling requirements, depending on the complexity and potential risk associated with the device.
- Indications for Use: A clear description of the intended use of the medical device.
- Contraindications: Any specific conditions or circumstances under which the device should not be used.
- Side Effects and Adverse Reactions: Information on possible side effects or adverse reactions associated with the use of the device.
- Special Precautions: Any special precautions that need to be taken during use.
- Clinical Data and Performance Information: For higher-risk devices, clinical data, and performance information might be required to support safety and efficacy claims.
Also Read: Class II & III Medical Device Registration in Indonesia
Clinical Trial Requirements
Clinical Trial Authorization: Before initiating a clinical trial for a medical device in Indonesia, the sponsor (usually the device manufacturer or its representative) is required to obtain approval from BPOM. Documents required while applying to the BPOM includes Clinical trial documents, Clinical Trial Protocol, Informed Consent, Form Appendix, Product documentation, and Product information such as Certificate of Analysis (CoA), Certificate of GMP, and Summary Batch Protocol.
Ethical Approval: In addition to BPOM’s approval, the clinical trial must also receive ethical clearance from an accredited ethics committee in Indonesia. The ethics committee ensures that the trial protocol adheres to ethical principles, protects the rights and welfare of study participants, and complies with international guidelines for human research.
GCP Compliance: Clinical trials for medical devices in Indonesia must be conducted in accordance with Good Clinical Practice (GCP) guidelines, which outline internationally accepted ethical and scientific quality standards for designing, conducting, and reporting clinical trials.
Registration of Clinical Trials: BPOM may require the registration of clinical trials in a publicly accessible database, Indonesia Clinical Research Registry, providing transparency and facilitating the dissemination of information on ongoing and completed trials.
Product Testing Requirements
Performance Testing: Medical devices may undergo performance testing to evaluate whether they meet the intended purpose and demonstrate the required functionality accurately. This can involve tests on accuracy, precision, sensitivity, specificity, and other relevant performance parameters.
Safety Testing: Safety testing is essential to assess potential risks associated with the use of the medical device. This can include biocompatibility testing to determine the device’s interactions with living tissues, material safety assessments, electrical safety testing, and other evaluations to ensure that the device does not pose undue risks to patients or users.
Labeling and Packaging Testing: Testing of labels and packaging materials is often required to verify that they meet the necessary standards, provide essential information to users, and maintain the device’s sterility, if applicable.
Sterilization Validation: Medical devices intended for sterile use may need to undergo sterilization validation testing to ensure that the chosen sterilization method effectively eliminates microorganisms while preserving the device’s integrity.
Software Verification and Validation: For medical devices with software components, verification, and validation testing may be required to ensure the software’s reliability, security, and compliance with relevant standards.
Biocompatibility Testing: Certain medical devices that come into direct or indirect contact with the human body may need to undergo biocompatibility testing to assess their potential for irritation, sensitization, cytotoxicity, and other biocompatibility aspects.
Shelf Life and Stability Testing: For medical devices with specified shelf life, stability testing may be necessary to determine the product’s stability under various storage conditions over time.
As per Medical device Regulations, Registration in Indonesia requires ISO 13485 certificates for attestation of the quality system of the manufacturing facilities.
Medical Device Registration Process in Indonesia
Regulatory Liaison: The local representative acts as the intermediary between the foreign manufacturer and BPOM. They are responsible for submitting the necessary documentation and information to BPOM on behalf of the manufacturer.
Important Documentation: The manufacturer or the authorized representative of the medical device in Indonesia is responsible for preparing the required documentation. This typically includes technical and administrative documents such as
- Executive Summary
- Device Labelling
- Detailed Manufacturer Information
- ISO Certificate for device conformity
- Summary of Design verification and validation documents
- Risk Analysis
- Method of destruction
- Quality Management Certification
- Letter of Certification of the intended use/indication/ Package, Letter of certification of label and instruction for use from manufacturer or product owners
- Declaration of Conformity
- Letter of Confirmation for market history proof from manufacturer
- Letter of Safety confirmation from manufacturer
- Proof of approval from Medical Device Regulatory Authority in Foreign Countries
- Letter of Authorization
- Grouping indication letter (if needed)
Submission of Application: Once all the necessary documentation is prepared, the application for medical device registration is submitted to the BPOM. The application should be completed accurately and include all required information to avoid delays in processing.
Review and Evaluation: The BOPM reviews the submitted documents to assess the device’s safety, quality, and conformity with applicable regulations and standards.
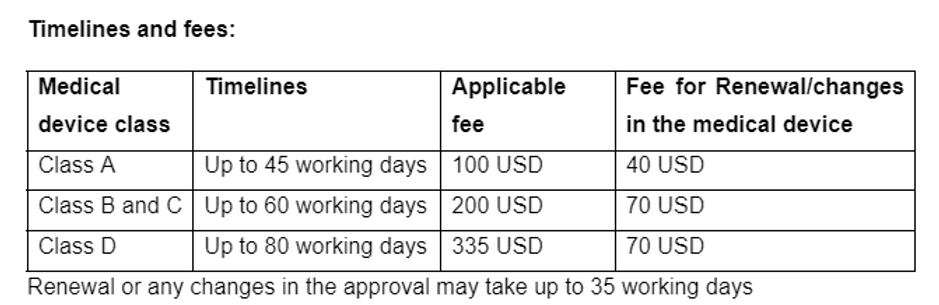
License Validity: All product licenses are valid for a period of two – five years, depending on the validity period of the Letter of Authorization.
Import and Distribution Requirements
Before importing a medical device into the country, products should have a valid Distribution Approval Number or NIE (Nomor Izin Edar) otherwise known as the Product Approval License from the Ministry of Health via the Online Medical Device Registration platform.
Only the local Indonesian representatives can apply for the NIE and the IDAK (via the OSS) for registration and importation of medical devices.
A medical device can only be registered by one IDAK (distributor license holder) at a time. Multiple registrations of medical devices with different companies or distributor license holders are not allowed in Indonesia.
Good Method of Distribution of Medical Devices (or CDAKB) is implemented in order to enforce good distribution practice requirements in the country which requires manufacturers to have:
- Quality management system and Resource management
- Building and facilities & Storage and inventory handling
- Product traceability
- Complaint handling
- Field safety corrective action
- Call back and Destruction system
- Illegal and unqualified medical devices recording
- Internal audit
- Management review
- Outsourcing activity (if required)
The approval or certification from reputable regulatory authorities such as the USFDA or the EMA can provide valuable supporting evidence of the device’s safety, quality, and efficacy. Prior approval can streamline and expedite the registration process in Indonesia. However, it’s essential to note that the approval or certification in the EU or US does not automatically guarantee acceptance by the Indonesian National Agency of Drug and Food Control (BPOM), however, it can fast-track the process:
Expedited Review: BPOM may consider regulatory approval from the EU or US as part of its review process, potentially expediting the evaluation of the medical device in Indonesia.
Technical Documentation and Clinical Data: The technical documentation and clinical data submitted for approval in the EU or US can be used to support the registration application in Indonesia. This may reduce the need for duplicative testing or documentation.
Regulatory Compliance: Regulatory approval in the EU or US indicates that the medical device meets the necessary standards and regulations in those regions, which may be considered favorably by BPOM during its assessment.
GMP Inspection Requirements:
Inspection: BPOM conducts inspection in two ways, with pre-notification or without pre-notification depending on the history of product recall and if the inspection is not found satisfactory up on pre-notified inspection. During a GMP inspection, BPOM assesses various aspects of the manufacturing facility, including the production processes, quality control measures, equipment calibration, personnel training, and record-keeping practices.
Risk-Based Inspections: The frequency and intensity of GMP inspections may be determined based on the risk classification of the medical devices being manufactured. Higher-risk devices or facilities with a history of compliance issues may be subject to more frequent inspections.
Pre-Approval Inspection: GMP inspections may be conducted as part of the pre-approval process for new medical devices seeking registration in Indonesia. This inspection verifies that the manufacturing facility meets the required standards before the device is allowed to be marketed in the country.
Post-Market Inspections: Even after a medical device is registered and marketed in Indonesia, BPOM may continue to conduct periodic post-market GMP inspections to ensure ongoing compliance and product quality.
Corrective Actions: If any non-compliance or deficiencies are identified during the GMP inspection, the manufacturer is typically required to take corrective actions to address the issues. BPOM may reinspect the facility to verify that the corrective actions have been implemented satisfactorily.
Post Marketing Surveillance
Adverse Event Reporting: Manufacturers, importers, and distributors of medical devices are required to report adverse events and incidents related to their devices to the BPOM. All adverse events should be reported:
- Within 48 hours for events that represent a serious threat to public health;
- within 10 days for events that led to death or serious deterioration in the state of health and;
- within 30 days where multiple events which have led to death or serious deterioration in the state of health.
- And Periodic Safety Updates are required to be submitted to BPOM
Field Safety Corrective Actions (FSCA): If a safety issue or device malfunction is identified, manufacturers may need to implement field safety corrective actions, such as product recalls or product modifications, to address the problem and protect patients and users in accordance to Medical Devices Regulations in Indonesia
References:
- https://ina-registry.org/index.php?act=news&id=6
- https://pubmed.ncbi.nlm.nih.gov/7552240/