Hong Kong- New Drugs, Generics, Biosimilar Regulations: Hong Kong has a robust regulatory system for pharmaceuticals, overseen by the Department of Health (DoH). This system prioritizes the safety, efficacy, and quality of medicines (New drugs, Generics, Biosimilar) available to the public. Here’s a breakdown of the key aspects of regulations and process:
New Drugs, Generics, Biosimilar Regulatory Framework In Hong Kong
- Pharmacy and Poisons Ordinance (Cap. 138): This is the cornerstone legislation governing pharmaceutical regulations in Hong Kong.
- Pharmacy and Poisons Regulations (Cap. 138A): These regulations provide detailed requirements for registration, labeling, advertising, and other aspects of pharmaceutical products.
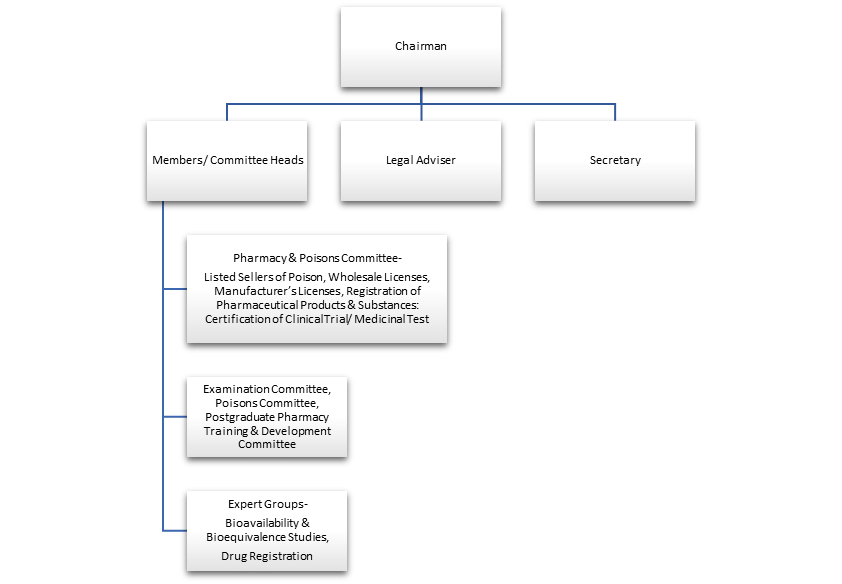
The Role of the Department of Health (DH):
Drug Office: This department within the DH is responsible for implementing the Pharmacy and Poisons Ordinance and its regulations. They handle tasks like:
Registration: All pharmaceutical products must be registered with the Pharmacy and Poisons Board (PPB) before being sold in Hong Kong.
Inspections: The DH conducts inspections of manufacturing facilities and distributors to ensure compliance with Good Manufacturing Practices (GMP).
Pharmacovigilance: They monitor the safety of registered drugs after they reach the market and investigate any adverse drug reactions.
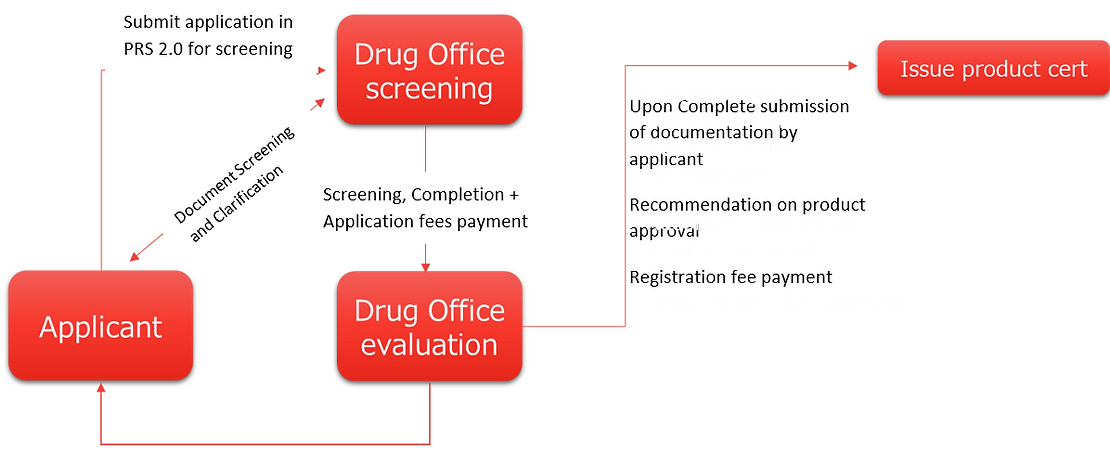
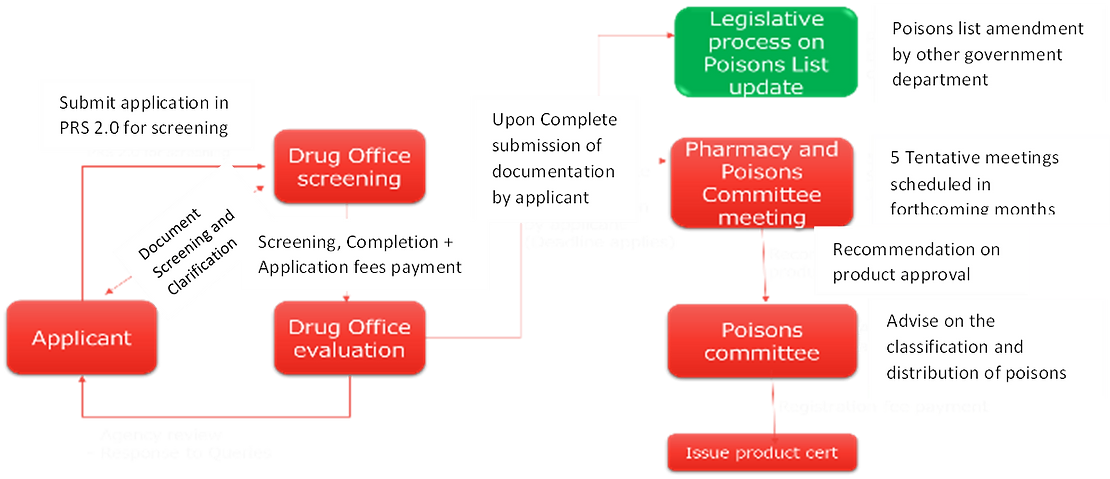
Registration Process of New Drugs, Generics & Biosimilar in Hong Kong
- Manufacturers are required to submit a detailed application package to the PPB, including data on the product’s safety, efficacy, and quality control procedures.
- The PPB reviews the application and may request additional information before granting registration.
- Upon Complete submission of documentation by applicant
- Recommendation on product approval
- Registration fee payment
- Once registered, a unique registration number is assigned to the product, which must be displayed on the packaging.
- Screening, Completion + Application fees payment
- Document Screening and Clarification
- Submit application in PRS 2.0 for screening
Below Mentioned is the registration process for Generics and Biosimilar in Hong Kong
New Drugs, Generics and Biosimilar Products Timelines In Hong Kong:
- Screening phase ~2-3 weeks;
- Generic Evaluation ~ 6-12months subject to complexity and queries response time;
- Biosimilar ~ 12-18 months subject to complexity and queries response time
List of documents for New Drugs and Generics(Hong Kong)
(*both Electronic copy and original/certified true copies are required):
- Copy* of Manufacturer(s)’ licence (from all mfr involved in the DP production)
- Copy* of GMP cert of the mfr(s) with evidence of PIC/S GMP standards
- Overseas manufacturer’s SMF or equivalent
- Copy* of FSC or CPP of the product issued by COO
- Master Formula
- Release specification and Shelf-life specification of the finished product
- Method of analysis of the product for all tests
- CoA of the finished product
- Stability test data (at least 6m accelerated + on-going real time stability data)
- Prototype sales pack and proposed package insert
- Reputable reference/approved PI as supporting documents
- Color photos or scanned image of the product
- Clinical and scientific documentation substantiation the safety and efficacy
- Bioequivalence data
Additional List of documents required for Biosimilar
- Evidence to support the product as a Biosimilar to the reference product, additional support if differences are found
- At least 1 MA from one of the reference agencies and its FSC – EMA/ MHLW/ TGA/ Health Canada
- Quality Documents
- Full quality dossier : Separate section on comparability exercise, Justification for any observed differences w.r.t their potential impact on safety and efficacy, Requirements for the amount of non-clinical and clinical data considered as case-by-case approach depending the differences of the quality data
- For reduced non-clinical and clinical data, full quality dossier (including comparability exercise) is required
- Non-Clinical Documents: 1) in vitro studies, 2) in vivo studies, 3) Other toxicological studies when necessary
- Clinical Documents: 1) Clinical data of the BS product between the BS and reference products (i.e. PK studies, PD studies, clinical efficacy and safety trials), 2) Clinical studies demonstrate clinical comparability between BS and reference products, 3) efficacy/equivalence clinical studies (Any extrapolation needs to be scientifically justified)
- Immunogenicity Studies
- Pharmacovigilance requirements
ADR reporting
PSURs submission for 5 years at a regular basis
RMP and/or REMS by reference countries
Educational materials for HCP on specific risks of the BS products and measures to reduce them
package inserts for patients
Specific labelling requirements for BS
Registration Process for New Chemical Entity or Biologic
Timelines:
- Screening phase ~2-3 weeks;
- Generic Evaluation ~ 5- 12 months subject to complexity and queries response time
Additional List of documents required for New Chemical/ Biological Entity:
- CPP from two or more of the reference countries
- ICH CTD M2 (or equivalent) , M3 and M5 (if appropriate)
- Evaluation reports on safety, efficacy and quality of the product signed by expert(s)
- Expert’s CV
- RMP/ REMS to be implemented in HK, with reference to the RMP required by the reference countries
- Risk assessment report of elemental impurities
- Information of any pre-registration impartation (e.g. NPP and clinical trials)
- Comparison of indications, dosage, warning/precautions, contraindications or side effects in other countries
- Worldwide registration status
- Any refused/suspended/ revoked product authorization in ref. countries
Post-Market Surveillance: Pharmaceutical companies must conduct post-market surveillance to monitor the safety and efficacy of their products once they are on the market. This involves reporting adverse events, conducting pharmacovigilance activities, and complying with DoH’s requirements for post-market monitoring.
Pricing and Timelines: The timeline for New Drug submissions is approx., 8-15 months whereas for Generics it is 6- 12 months and Biosimilar it is 12-18 months. The DoH fees varies for New Drug, Generics and Biosimilars, is USD 315
There are different registration requirements for various types of medicines, such as Western medicines, Chinese medicines, and controlled drugs.
Understanding these intricacies of regulatory affairs in Hong Kong is crucial for pharmaceutical companies and distributors to ensure their products meet the required standards and contribute to a safe and effective healthcare environment.

