
China being one of the world’s fastest growing pharmaceutical markets has a comprehensive regulatory framework to keep up with the rapid advancement of the technological and scientific aspects of the research and development system. The framework allows for the safe, effective and quality production as well as marketing of pharmaceuticals. The China Food and Drug Administration (CFDA) was founded in 2013 and was responsible for the regulation of drugs, medical devices, food, cosmetics. The CFDA was a ministerial level agency that was under the State Council.
Later, as part of China’s Government Administration restructuring, The CFDA was transformed into the present National Medical Products Administration (NMPA). There was a time where the public health needs were affected by the slow pace of the drug approval system in China. So , to counteract this, the NMPA has implemented several accelerated pathways for the drug approval process. Read below to know more about these pathways and the pharmaceutical regulations in China.
Pharma Regulatory Bodies In China
National Medical Products Administration (NMPA):
NMPA is the main regulatory body in China which handles pharmaceutical drug registration management, look after formulating and organizing drug registration, review them and approve or reject them accordingly.
Centre for Drug Evaluation(CDE):
Under NMPA, Reviewing the clinical trials of drugs, applications for drug marketing authorization, any supplementary applications and applications for re-registration are done by CDE.

Classification Of Drugs In China
As per Pharmaceutical Regulations in China, NMPA classifies drugs into 3 categories and on the basis of these categories the products are developed or registered, those categories are-
1. Chemical Drugs – 6 classes (NCE Related -2)
2. Biological Drugs – 15 classes
3. Traditional Chinese medicines – 9 Classes
They are also classified into 5 categories for drug application as-
1. Improved new drugs
2. Innovative drugs
3. Import drugs
4. Generic drug (Original Drug approved in China)
5. Domestic generic drug (Original drug not approved in China)
There are 2 registration path-way for foreign manufacturers that include improved new/innovative drug or imported drugs and 2 clinical strategies which include parallel development and sequential development.
All the foreign drugs that belongs outside China are required to register as DMFs via Imported Drug application.
For New drug applications (NDA) NMPA has initiated 4 programs for accelerating the review and approval of drug which includes-
a) Conditional approval (CA): This approval procedure aims to shorten the research and development time for the clinical trials of the drug. This is done to render drugs as early as possible for patients who no longer can wait with a critical illness they have.
b) Breakthrough therapy designation (BTD): This procedure is for the drugs which are used to prevent and/or treat life threatening diseases or diseases seriously affecting the quality of life of the patients.
c) Priority review (PR): The priority review procedure is for Marketing Authorization Applications (MAA). With priority review of applications, it can shorten the waiting time for the drug to be reviewed by the regulatory body for approval and thereby expediting market entry.
d) Special approval (SA): This is an accelerated approval procedure which is considered when there is a public health crisis or potential health emergencies, thereby aiming for the prevention and treatment of such emergencies. This process has been terminated in 2020.
NMPA and its institutions offers technology and policy support which includes communication, technical guidance allocation of resources and decreasing the review time to the applicant if NDA enters any of these categories.
For the registration of generic product for importers or overseas manufacturers without legal representation needs to apply for product registration through agent services. The application is submitted to CFDA after which CDE runs through review process of the product.
Documents Required For Drug Registration In China
- Dossier shall be directory as per the “Provisions of Drug registration” for application items.
- Document with the information: Name of application item or drug/product, item number, applicant name, address and contact number.
- Import License (Import drug registration application form)
- Approval Certificate
- Import quota certificate (wherever applicable)
- Inspection or Clinical testing certificate (where applicable)]
- Quality and safety license
Application For Clinical Trial
- Application for clinical trial is submitted
- CDE carry out the test report and overall documentation review which takes about 40-60 days depending on the product.
- The review report is further sent to CFDA with the recommendation by deciding that the product should opt for clinical trial or bioequivalence study.
- If it opt for clinical study it can be divided into 4 phases
- If applicant receives clinical trial or bioequivalence study approval, the applicant is free to choose the hospitals for conducting clinical trials from the given clinical trial hospitals list and all the study should be carried out in compliance with GCP (Goods Clinical Practice).
- After the successful completion of clinical study, Manufacturer/ applicant gets the approval document along with the consent form and study reports which forms the documents required for the drug registration application.
- SFDA Evaluation center will evaluate and review the information. If it passes, the file is passed for final approval and provides Pharmaceutical registration certificate which is valid for 5 Years.
Import Drug Registration Process As Per Pharmaceutical Regulations In China
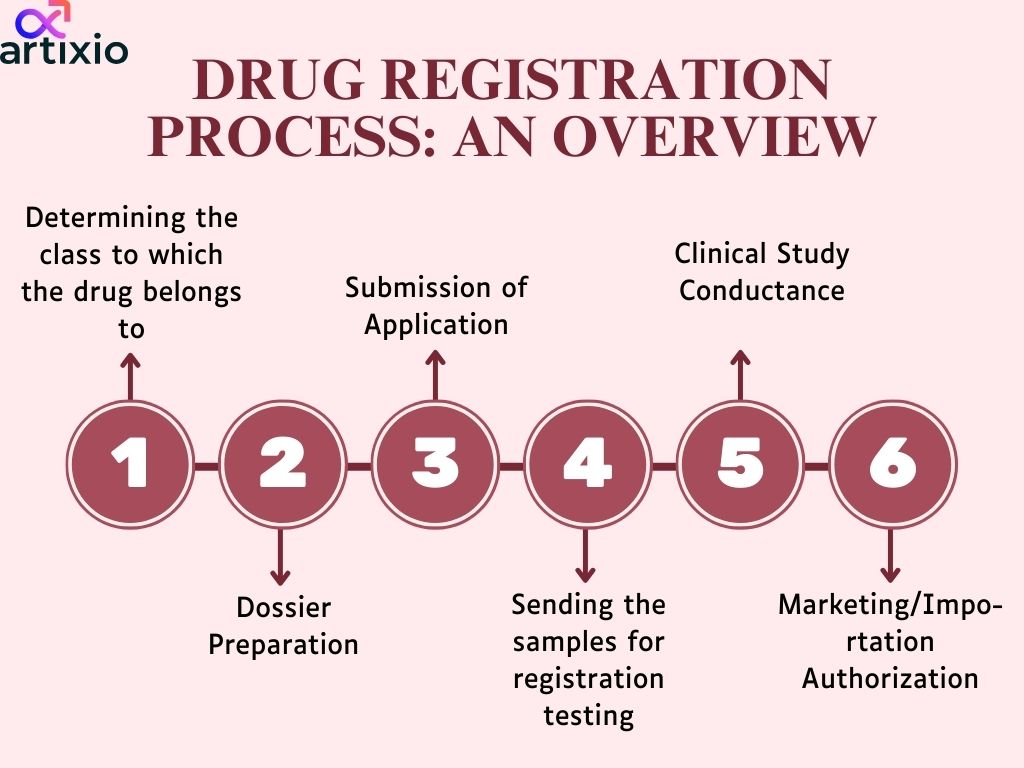
- Applicant submits the clinical study report and other document along with import drug registration form;
- CFDA/SFDA review evaluate and send notification if any additional information required;
- SFDA CDE evaluation takes about 120 days and if additional document requires then takes about 160 days;
- CDE review the drug in about 40-50 days and send report to SFDA/CFDA;
- SFDA/CFDA approval takes about 20-30 days. And the Import registration certificate is provided to the applicant or manufacturer/importer.
Timeline For Drug Registration
It takes about 8-9 months or 270- 350 days for full registration process which includes 30-35 days after the submission of application for notification, 80-90 days for evaluation of clinical trials, 30-40 days for approval by NMPA, 120-150 days for evaluation of drug by CDE and again 30-40 days to get the approval from NMPA after clinical trials.
Approval from the US/EU could fast-track the approval in China
China’s National Medical Products Administration (NMPA) does consider approvals from well-regulated international agencies like the FDA and EMA as part of the evaluation process. This is because the FDA and EMA have rigorous and comprehensive regulatory standards for drug approval. Therefore, if a drug has been approved by these agencies, it can provide valuable data and evidence supporting the drug’s safety, efficacy, and quality.
However, the NMPA still requires the applicant to provide additional data and information specific to the Chinese population and regulatory requirements. This includes data from Chinese clinical trials, safety and efficacy data in Chinese patients, and other relevant documentation.
The NMPA’s approval process for foreign drugs typically involves the following steps:
- Acceptance and Evaluation: The applicant submits the required data and documentation to the NMPA for evaluation. This includes data from foreign clinical trials and regulatory approvals.
- Bridging Studies: In some cases, the NMPA may request “bridging studies,” which are additional clinical trials or studies conducted in Chinese patients to confirm the safety and efficacy of the drug in the Chinese population.
- Technical Review: The NMPA conducts a technical review of the submitted data to ensure compliance with Chinese regulatory requirements and standards.
- On-Site Inspection: Depending on the product and other factors, the NMPA may conduct on-site inspections of the manufacturing facilities.
- Approval Decision: Based on the evaluation and review process, the NMPA makes a decision on whether to approve the drug for marketing in China.
Having approvals from reputable international agencies like the FDA or EMA can streamline the Chinese approval process as it provides a foundation of prior evaluation and a certain level of confidence in the drug’s safety and efficacy. However, it’s important to note that the NMPA’s approval process may still have its own unique requirements and considerations specific to the Chinese market.
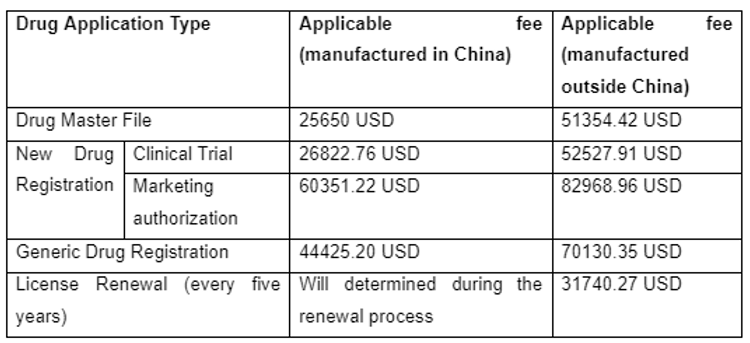
Drug Application Fees in China
Post Marketing Surveillance
The NMPA has established a robust pharmacovigilance system to gather, analyze, and evaluate data related to adverse drug reactions. This system enables the detection of any potential safety signals or emerging risks associated with drugs. The National Adverse Drug Reaction Monitoring Information Network serves as a key component of the pharmacovigilance system and plays a crucial role in monitoring and evaluating the safety of pharmaceutical products available in the Chinese market.
Risk Assessment and Evaluation: The NMPA conducts risk assessments and evaluations based on the reported adverse drug reactions and other safety data. This process helps in determining the severity and frequency of ADRs and in making decisions on the safety profiles of drugs.
Artixio can help you obtain the marketing authorization for pharmaceutical products in China using our regional teams. Please get in touch with us today for any assistance needed in China.
FAQ’s
Q. What are the responsibilities of NMPA?
NMPA is responsible for the supervision of the safety, regulation of the registration and in undertaking standards management for drugs, medical devices and cosmetics.
Q. Why was the Special approval procedure terminated?
A. The benefits of this procedure became less tangible after a formal avenue for communication was established in 2016 between the regulators and the sponsors. So, it was terminated in 2020 and was not included in the 2020 Drug Registration Regulation (DRR).
Q. What are the accelerated review pathways implemented by China?
A. There are 4 accelerated review pathways put in place by China. They are Conditional Approval, Priority Review, Breakthrough Therapy and the Special Approval pathway.
Q. When should supplementary application of confirmatory trial data be submitted for conditionally approved drugs?
A. The validity period of registration certificate for drugs approved via the conditional approval procedure is 5 years. 200 days is the review time for the supplementary application for conformity trial data. So, the supplementary application must be submitted within 4 years after the receival of conditional approval.

