
EU IVDR compliance requirements have become more stringent, with stricter conformity assessment expectations and comprehensive post-market surveillance obligations. All of this has been reshaped by the emergence of the EU In Vitro Diagnostic Regulation (IVDR). Reading further, you will get acquainted with the key changes from IVDD to IVDR, along with EU IVDR requirements explained in a practical way for IVD manufacturers seeking market devices in the European Union.
What is the EU IVDR?
The EU IVDR, Regulation (EU) 2017/746, is the current governing legislation for in vitro diagnostic devices within the EU and EFTA states. This regulation was published on 5 May 2017 and is brought into force since 26 May 2022. This IVDR replaced the previous IVD Directive (98/79/EC). For companies looking for in vitro diagnostic regulation EU requirements, IVDR provides a modernized framework designed to strengthen patient safety, device quality, and regulatory oversight.
Key Changes from IVDD to IVDR for IVD Manufacturers
IVDR is a lifecycle-based approach whereas IVDD is a device compliance specific approach.
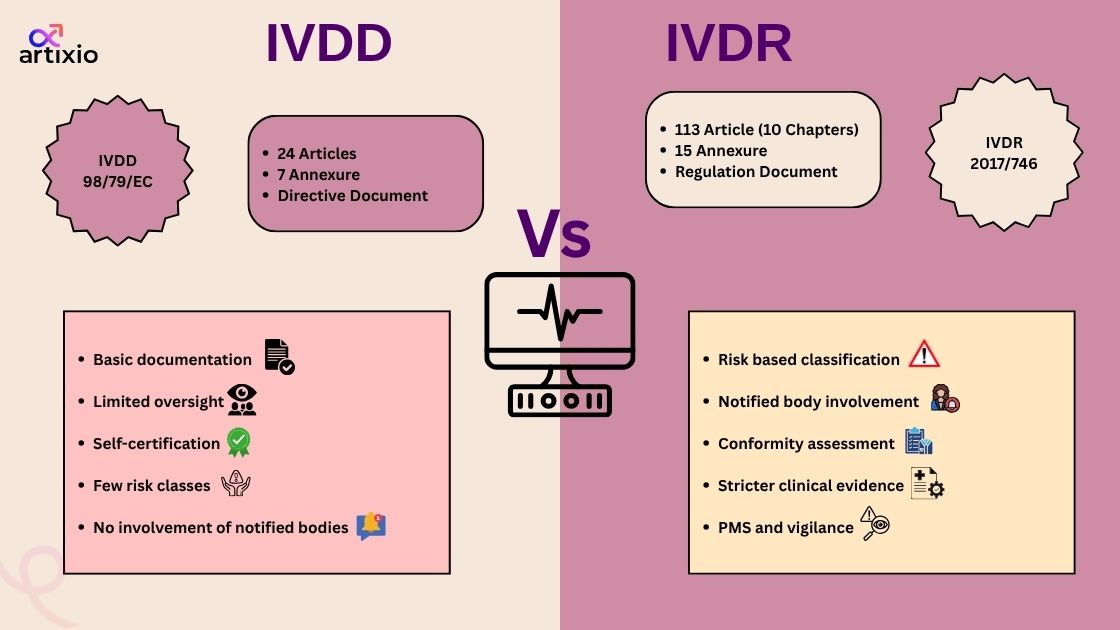
Below is detailed comparison on the key changes from IVDD to IVDR:
| Key Topic | IVDD (98/79/EC) |
IVDR (EU) 2017/746
|
| Legal Status | This was a directive which was published in 1998. This directive was required to be established at a national level. |
This is a regulation that was proposed in 2017. The IVDR applies to all EU member states.
|
| Validity Period | It was in forced until 25 May 2022 |
It came into force since 26 May 2022 and superseded the IVDR
|
| Scope | It covered reagents, calibrators, control materials, kits, instruments, specimen receptacle |
It extended its scope which included software such as IVD, companion diagnostics, and genetic tests.
|
| Risk Classification | For risk classification, it included two lists that is Annex II list A and B |
Here, the regulation aligned with international standards of risk classification that are class A, B, C, and D.
|
| Conformity Assessment | Under this directive, conformity assessment is the responsibility of the manufacturer, for high-risk devices notified bodies are also involved. |
Under these regulations, the conformity assessment is risk-based, and it depends upon the notified bodies for class B to D, with stringent oversight
|
| Documentation | Less prescriptive documentation is required. |
Detailed documentation is required.
|
| Role of Economic Operators | Here manufacturers are majorly focused. |
Clear obligations are stated for all that is manufacturers, importers, distributors, authorized representatives.
|
| Notified Bodies | The notified bodies are designated nationally with limited oversight. |
The notified bodies get stricter designation, EU-level monitoring, unannounced audits, expert panels for Class D
|
| Clinical Evidence | Less stringent requirements for clinical evidence. |
In detail, clinical evidence is required such as Performance evaluation report (PER) and performance studies required.
|
| Post-Marketing Surveillance | A less detailed post-marketing surveillance is required. |
A detailed post-market performance follow-up (PMPF), vigilance, and periodic safety update reports (PSUR) are required for PMS.
|
| Person Responsible for Regulatory Compliance (PRRC) | Not required. |
It is compulsory to appoint a PRRC.
|
| Public Transparency | It was limited to the national level of reporting. |
Higher transparency is promoted with all data been published under the EU database.
|
| Global Harmonization | The IVDD did not focus much on international harmonization. |
The IVDR aligns with the IMDRF and global standards.
|
| EUDAMED Database | Not applicable. |
A centralized European database for medical devices, including UDI/device registration, economic operators, and vigilance data, is established under the IVDR.
|
EU IVDR Classification Rules for IVD Manufacturers
In the EU IVDR (Regulation 2017/746), the devices are clearly classified based on the risk associated with the device. The rules for the same are laid down in Annex VIII of the IVDR, which replaces the guidelines from the IVDD.
The IVDR basically classified a device into any one of the four categories that is class A, B, C and D. Apart from this, the IVDR states to follow the rules laid down in Annex VIII for the deciding the class of the device intending to be marketed.
There are seven rules mentioned in the Annex VIII for classification of IVDR, which are as follows:
| RULE NO | DESCRIPTION | EXAMPLE |
DEVICE CATEGORY
|
| Rule.no 1 | If the device is used to ensure safety of blood, organs, or tissues |
|
Class D |
| Rule no 2 | If the device is used to diagnose infections in patients |
|
Class C |
| Rule no 3 | Devices used for transfusion and transplant compatibility such as blood grouping or tissue typing |
|
Class D |
| Rule no 4 | Self-testing devices. For low risk within this category its class B and for high-risk device its class C |
|
Class B and C |
| Rule no 5 | Devices used as general laboratory devices |
|
Class A |
| Rule no 6 | Devices used for tests guiding treatment decisions that is for companion diagnostics |
|
Class C |
| Rule no 7 | Devices which are not categorized under any of the above-mentioned rules but are used for diagnostic purposes. |
|
Class B |
EU IVDR Compliance Requirements for IVD Manufacturers
Following are some of the key IVDR regulation requirements and practical compliance expectations for manufacturers operating under the EU IVDR:
- Risk-based classification: All the devices intending to regulate in the EU must classify their device into class A, B, C, and D. Higher classes may require notified body review.
- EUDAMED registration: The device should mandatorily register with the EUDAMED database.
- Notified Body: IVD devices that fall under class B, C, and D are required to notify body involvement for review of their technical documentation and performance evaluation.
- Conformity assessment: According to the risk classification of the device, the manufacturer must complete the applicable conformity assessment which often involves notified bodies.
- CE Marking: All the devices must possess the CE marking/symbol for successful conformity assessment for successful EU marketing.
- Technical documentation: A detailed documentation involving the process and product design, intended use, performance evaluation, and risk management should be submitted for the registration of the IVD.
- Performance evaluation: Evidence of scientific, analytical, and clinical performance of IVD should be submitted.
- Quality management System (QMS): The manufacturer must implement a QMS system aligning with the ISO 13485 throughout the IVD lifecycle procedures.
- Post-marketing surveillance and Vigilance reporting: Continuous monitoring and vigilance reporting for the IVD should be performed and submitted.
- Unique Device Identification (UDI): The device must mandatorily possess a UDI identification in the EUDAMED database to ensure its authenticity and traceability.
EU IVDR Regulations for IVDs Registration
It is essential for every IVD manufacturer in the EU to prepare an IVDR regulatory checklist to ensure that the device meets all key regulatory and documentation expectations. Below are some of the major checklist points to keep in mind while preparing such a checklist.
- Is the IVD correctly classified based on the risk category from Annex VIII rules
- Is each step of IVD product lifecycle compliant to ISO 13485
- Are all the documents prepared correctly in compliance with the Annex II and Annex III requirements
- Are the scientific, analytical and clinical performance of the device evaluated
- Is conformity assessment done for the device and if the device is of category B, C or D, are the documents submitted to the notified body
- Does the device possess CE marking
- Are the device and manufacturer registered on EUDAMED
- Does the label comply with the IVDR requirements such as UDI number, local language usage, CE marking, etc.
- Has the device well established post-marketing plan and process such as vigilance reporting, trend analyzing, PMS reporting, etc.
- Has there been well established stakeholder communication
EU IVDR Approval Process (Step-by-step):
IVDR is a lifecycle approach that emphasizes safety, performance, and continuous compliance. Through this approach, the EU ensures that each IVD available in the market meets all the stringent quality and regulatory standards. Below is a detailed step by step pathway that manufacturers must follow for their IVD’s approval:
- Device Classification:
Classify the IVD based on the risk associated with it according to Annex VIII. The classification of the device further decides if involvement of a notified body is required or not. - Quality Management System (QMS):
Establish and ISO 13485 compliant QMS system for the IVD product lifecycle that regulates all aspects such as design, production, risk analysis, performance evaluation, labeling, etc. - Technical Documentation Preparation:
Prepare and analyze all technical documents according to Annex II and III for the IVD registration. Documents included device description, intended use, risk analysis, performance evaluation, labelling, design analysis, etc. - Performance Evaluation:
A complete Performance Evaluation Report (PER) should be submitted for the device registration that demonstrates scientific validity, analytical performance, clinical performance, etc. - Conformity Assessment:
If the IVD falls under any category except for class A, it is liable for conformity assessment by the notified body whereas for class A self-declaration of the conformity assessment is required. - CE Marking:
After the approval of conformity assessment by the notified bodies for class B, C and D and self-certification for class A, the manufacturer should add a CE marking, which is a kind of symbol on the device and its packaging. This symbolizes EU IVDR compliance standards. - UDI and EUDAMED Registration:
The device should be assigned as a Unique Device Identifier (UDI), for the traceability and registration of the device, manufacturer, and economic operators in the EUDAMED database. - Authorized Representative Appointment (If Non-EU Manufacturer):
For all non-EU manufacturers, a local authorized representative must be appointed to carry out all the regulatory operations in the EU. - Post-Marketing Surveillance (PMS):
A stringent PMS system should be incorporated to track the device performance to keep it compliant to the EU IVDR requirements and ensure its safety for the users. Reports such as Periodic Safety Update Reports (PSUR). Post-Marketing Performance Follow-Up (PMPF), etc. should be submitted to ensure ongoing safety of the IVD. - Continuous Device Compliance:
Compliance should not come to an end once the IVD gets approved for marketing, but the actual compliance begins then. Manufacturers must ensure and keep updating the status and reports for IVD compliance through its lifecycle.
IVDR Transition Timelines
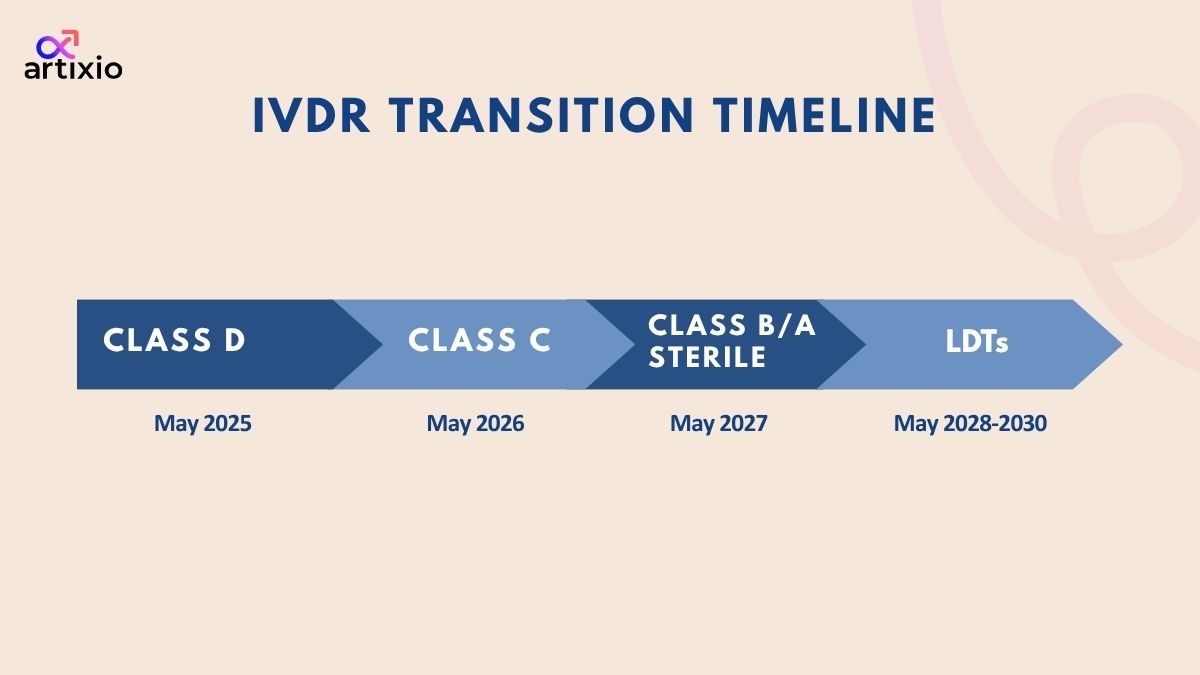
- IVDR was published on 5th of May and came into force on 26th of May 2017.
- To avoid disruption in the system, the EU provided transitional provision for compliance depending on the category of IVDR.
- Some of the transitional deadlines according to the type off device are as follows:
| Device Type |
Transitional Deadline
|
| Class D device | Until May 2025 |
| Class C device | Until May 2026 |
| Class A and B sterile devices | Until May 2027 |
| Lab Developed Tests (LDTs) | Until 2028-2030 |
- Other than these device categories, the certificate under IVDD remains valid until its expiry but not after May 2025.
- EUDAMED modules are slowly and gradually getting integrated with their mandatory use only when fully functional.
IVDR Regulatory Strategy Planning
A thorough strategy is required for the transition from IVDD to IVDR. This is especially important for teams interpreting IVDR requirements for IVD manufacturers and building a practical roadmap for compliance. A well-planned regulatory strategy helps manufacturers achieve complete alignment with IVDR principles. Following are some of the key strategy points to consider while complying with the IVDR:
- Gap Assessment: Compare the IVDR with the IVDD. Check with all the new requirements and prepare and proper gap assessment report addressing all the gaps and strategies to address those gaps.
- Focus on High-Risk Devices: As the IVDR has stringent rules for high-risk devices, their criteria for compliance should be prioritized first.
- Engage Notified Bodies: The capacity with notified bodies is limited, so the manufacturer should approach the notified bodies without any delays.
- Implement QMS: Implement ISO 13485 and IVDR Article 10 requirements throughout the device lifecycle.
- Register with EUDAMED: Prepare and register the device and manufacturer with the EUDAMED database.
- Train and Communicate: The complete organizational team should be trained and informed about each of the parameters and new requirements of the IVDR.
Conclusion:
Thus, we can conclude that the emergence of IVDR has introduced a complete device lifecycle approach. This leads to harmonization with the international requirements for IVD and a better developed device in terms of quality, efficacy, and patient health. IVD Manufacturers who are intending to enter the EU market or are already established should prepare a thorough plan and checklist to ensure compliance with the IVDR requirements.
We at Artixio, with a well-experienced and qualified local regional team, can help ensure your IVD’s complete compliance with new IVDR requirements. Our team supports manufacturers with EU IVDR requirements explained in a practical, implementation-focused manner, helping you navigate complex regulations and achieve timely compliance for successful market placement in the EU.
For more details, contact at info@artixio.com
FAQs:
1. Is it mandatory for class A non-sterile devices to have CE marking on their device?
Yes, although class A non-sterile devices do not require involvement of notified bodies but are liable to submission of self-conformity assessment of their device and CE marking.
2. What are the key points to check on the label for IVDR compliance?
The label should consist of CE marking, UDI identification, importer details, and instructions in local language for IVDR label compliance.
3. What should be the process for LDTs (Lab Developed Tests) for IVDR compliance transition?
Labs should comply with the IVDR requirements and should be used under transitional arrangements until 2028-30.
4. Can the client continue using IVDD certified devices stock after May 2025?
No, after May 2025 the IVDD certificate expires, so devices that have already been placed in the market before this date can only be marketed.

