
Japan is one of the most regulated healthcare markets globally, and medical device regulations in Japan reflect that. No medical device can enter the Japanese market without meeting the requirements formed under the PMD Act. It influences classification decisions and review expectations. Even post-market compliance requirements trace back to it.
From an agency perspective, an unclear classification can trigger additional questions and an extended timeline for reviews. So, before exploring the registration pathways, a clear understanding of how Japan classified medical devices and which authorities are involved is essential.
In this article, we explain the Japan Medical Device Classification system, Japan Medical Device Registration Process, PMDA regulatory requirements, approval pathways, and key compliance requirements for market entry.
Regulatory Authority for Medical Devices in Japan
Medical devices in Japan are regulated by two key authorities.
- Ministry of Health, Labour and Welfare (MHLW)
- Pharmaceuticals and Medical Devices Agency (PMDA)
Regulatory policies for medical devices in Japan are established by MHLW. The ministry also makes the final approval decision for Class III and Class IV devices that require ministerial authorization.
PMDA is responsible for the technical review of medical device applications. It is also a primary contact point for higher risk device submissions, non standard Class II applications, foreign site registration and regulatory consultations.
RCBs are private certification bodies authorized by the MHLW to certify Class II medical devices that fall within the defined certification standard (Ninteikijun).
Japan Medical Device Classification
Medical device classification in Japan uses four classes divided under a risk based system established under the PMD Act. The system is aligned with the IMDRF principle. Each device is assigned a Japan Medical Device Nomenclature (JNMD) code. Together with the device’s intended use and applicable classification rules, the code helps determine the class.
This classification helps determine the applicable regulatory requirements and the Japan Medical Device Registration Process that manufacturers must follow.
| Class | Risk | Devices | Pathway |
| Class I | Low | Stethoscopes, Surgical Gloves | Notification |
| Class II | Low to Moderate | X ray films, Infusion pump |
Certification or Approval
|
| Class III | Moderate to High | Dialysis equipment, Orthopedic implants | PMDA Approval |
| Class IV | High | Pacemakers, Heart valves | PMDA Approval |
Class II is where things get more tricky for many manufacturers. Standard Class II devices with established certification standards go through third party certification. Non-standard Class II devices without applicable certification standards may require full PMDA Medical Device Registration review and approval.
Among the most common causes of registration delays in Japan are classification disputes. Based on our experience, they regularly rank in the top three.
MAH Requirements for Medical Devices
A Marketing Authorization Holder (MAH) is a licensed entity based in Japan that assumes legal responsibility for a medical device placed in the Japanese market. Since foreign manufacturers cannot directly register medical devices in Japan, appointing an MHA is a mandatory regulatory requirement.
Acting as the primary regulatory contact, the MAH manages submissions to PMDA and MHLW. Responsibilities also extend to quality oversight, post-market surveillance, adverse event reporting and product recall management when required.
Medical Device Regulatory Guidelines in Japan
Core guidelines for medical devices are:
- PMD Act (Act no. 145,1969; revised 2014)
- QMS Ordinance (Ministerial Ordinance No. 169 of 2004; updated 2021)
- GVP Ordinance (Ministerial Ordinance No. 135)
- PMDA Guidance Documents (Tsuchi)
- MHLW Certification Standards (Ninteikijun)
- IMDRF/GHTF Guidelines (adopted)
Reading only the PMD Act is a mistake manufacturers make more often.
Medical Device Registration Process in Japan
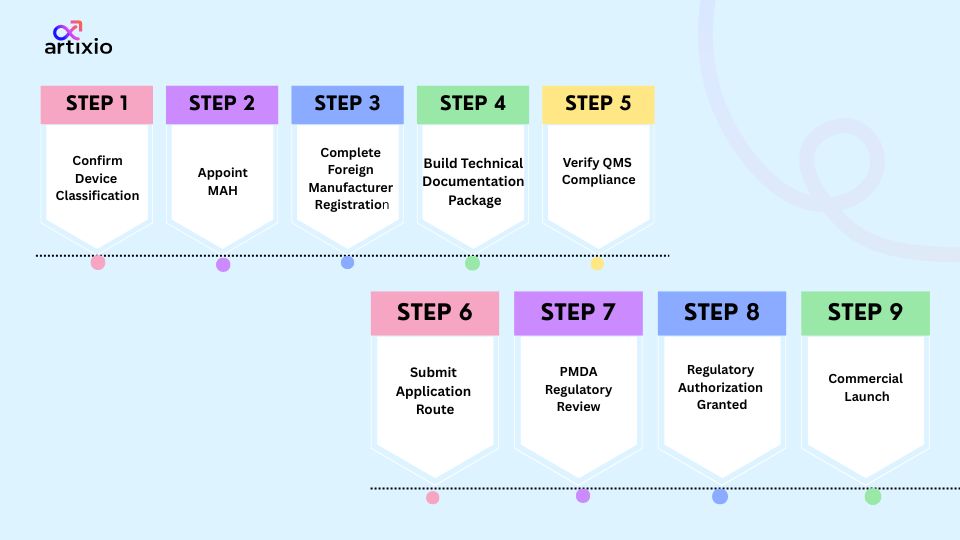
The medical device approval process in Japan follows a structured sequence. Here is the step by step approval process.
- Step 1: Confirm device Classification. Under Japanese medical device regulations, classification determines everything including the pathway.
- Step 2: Appoint a licensed Marketing Authorization Holder (MAH) in Japan. This is not a parallel task. It is a prerequisite.
- Step 3: Complete Foreign Manufacturer registration. Every overseas manufacturing site involved in production must be registered before certification or approval can move forward.
- Step 4: Build the technical documentation package. Device description, intended use, risk analysis, performance testing reports, and clinical data where required
- Step 5: Verify compliance with Japan’s Quality Management System (QMS). Run this in parallel. Compliance with Ministerial Ordinance No. 169 needs to be demonstrated before submission.
- Step 6: Submit the appropriate application route (notification, certification or approval)
- Step 7: PMDA regulatory review. Depending on the device classification and pathway, the PMDA may conduct a thorough assessment.
- Step 8: Regulatory authorization granted. It is issued following a successful assessment by PMDA and final approval from MHLW.
- Step 9: Commercial launch. Importation, distribution and market entry can begin once authorization is in hand and all labeling and import requirements are confirmed.
Among all stages, the preparation of technical documentation often has the greatest impact on review timelines. In complete or inconsistent technical files, gaps between the risk analysis and the clinical evidence. These are the most common reasons for delays during PMDA evaluation.
Required Documents for Medical Device Registration in Japan
Typical documentation includes:
Administrative Documents
- Application forms
- MAH information
- Foreign Manufacturer Registration certificates
Technical Documentation
- Device description
- Intended use statement
- Product specifications
- Design documentation
- Risk management file
- Verification and validation reports
- Sterilization validation (if applicable)
- Biocompatibility studies
- Software validation documentation
Clinical Documentation
Where required:
- Clinical evaluation reports
- Clinical investigation data
- Literature reviews
Quality Documentation
- ISO 13485 certificates
- QMS documentation
- Manufacturing process information
Japan QMS Requirements for Medical Devices
Japan’s Quality Management System (QMS) requirements are based on ISO 13485 but are governed by Ministerial Ordinance No. 169, which introduces country-specific requirements for medical device manufacturers. As a result, ISO 13485 certification alone does not ensure compliance with Japanese regulations.
What needs to be documented:
| Japan QMS Requirement |
ISO 13485 Reference
|
| Design and Development Controls | Clause 7.3 |
| Supplier and Component Control | Clause 7.4 |
| Sterilization and Process Validation |
Clause 7.5.6 and 7.5.7
|
| Complaint Handling and Post- Market Feedback |
Clause 8.2.1, 8.2.2, 8.2.3
|
| Document and Record Control | Clause 4.2 |
| Risk Management | Clause 7.1
and ISO 14971 |
Before approval or certification is granted, PMDA or Registered Certification Bodies (RCBs) review the manufacturer’s QMS through inspections and document assessments as part of the Medical Device Approval Process in Japan. This is not a formality. Any gaps identified can significantly delay the registration process.
What Artixio has seen repeatedly is that companies that align their QMS documentation with Japanese requirements early in the project typically experience a smoother review process. Manufacturers trying to bridge compliance gaps at the final stage always regret.
Medical Device Labeling & IFU Requirements in Japan
Every medical device marketed domestically in Japan must carry labeling and Instruction For Use in Japanese. This applies broadly across device categories and there is no exemption for imported products or devices originally developed for other markets. Translating existing labeling is not enough either. The content itself must meet Japan’s specific requirements, not just the language.
Product labels must include:
- Product name
- Device classification
- Approval and Certification number
- Marketing Authorization Holder (MAH) information
- lot or serial number
- Storage conditions
- Expiry date (when applicable)
The IFU serves a broader purpose. It needs to clearly cover intended use, contradictions, warnings, precaution, operating instructions, maintenance requirements and disposal guidance.
Depending on the nature and risk profile of the device, additional safety-related information may also be required to support proper use by healthcare professionals or patients.
What Artixio consistently sees in practice is labeling deficiencies remain one of the most common reasons regulators request clarifications or corrections. Addressing labeling requirements early in the Japan Medical Device Registration Process can help reduce review delays and support a smoother submission pathway.
Registration Timelines for Medical Devices in Japan
A Class I notification moves quickly. A class IV PMDA approval does not and no amount of internal pressure changes that. Device risk and pathway all influence how long the process actually takes.
| Device Type | Registration Route | Typical Timeline |
| Class I | Notification (Todokede) | 2-4 weeks |
| Class II (with Ninteikijun) | RCB Certification (Ninsho) | 2-6 months |
| Class II (without applicable standards) | PMDA Review (Shonin) | 6-12 months |
| Class III | PMDA Review (Shonin) | 6-18 months |
| Class IV | PMDA Review (Shonin) | 12-24 months |
| SAKIGAKE Designation | Expedited Review | Case dependent |
PMDA can issue additional questions mid review which can add time. Timelines may also vary based on product complexity, documentation quality.
SAKIGAKE Designation is worth flagging separately. It is Japan’s accelerated review program for innovative medical products, including certain medical devices, pharmaceuticals and regenerative medicine products. Not every device qualifies but for those that do it changes the timeline significantly.
Medical Device Registration Costs in Japan
Registration costs vary based on several factors. These include device classification, PMDA review fees, certification body fees, QMS inspection costs, translation expenses, MAH service fees and clinical evaluation requirements. Each element can affect the overall budget differently depending on the registration pathway.
Class III and Class IV registration cost more. Significantly more in most cases. The review is deeper, the documentation requirements are heavier. Inspection involvement is also greater.
Medical Device Import Requirements in Japan
Foreign manufacturers do not import directly. That is not how Japan works. All importation runs through the MAH. They bear full responsibility, hold the product approval or classification, act as the importer of record, and carry full responsibility for customs and regulatory import compliance.
A few things must be in order before any commercial shipment enters Japan:
- PMDA registration of the foreign manufacturing site
- Valid approval or certification number referenced in customs documentation
- Japanese-language labeling on all devices before market placement
- Lot and volume tracking maintained by the MAH for distribution traceability
Japan Customs enforces import controls in coordination with MHLW. devices arriving without proper documentation get detained. Sometimes returned. Neither outcome is quick to resolve.
Post-Market Surveillance Requirements in Japan
Japan’s post market surveillance framework is governed by GVP ordinance (Good Vigilance Practice). The MAH carries primary responsibility for everything that follows commercialization. All of it.
Core obligations:
- Serious adverse events reported to PMDA within 15 days. Other reportable events within 30 days
- Field Safety Corrective Actions and recalls managed and reported by the MAH
- Periodic Safety Update Reports required for designated high risk devices.
- Post market clinic follow-up where specified in approval conditions
- Complaint must be documented and investigated
- Annual distribution volume and safety reporting to MHLW
- QMS periodic review incorporating post-market data
- Labeling updates when safety information changes, with PMDA notification or approval required.
PMDA publishes detailed guidance on reporting timelines and event categorization. The MAH must have operational systems ready to receive safety signals from the foreign manufacturers. Then they need to assess them under Japanese requirements and report within mandated timeframes.
Conclusion
Japan is worth it. But only if approached correctly. The PMD Act framework is rigorous. PMDA’s review process is science based and thorough, and it does not move faster because a manufacturer’s internal deadline demands it. What makes Japan work for foreign manufacturers is preparation. Accurate classification, a capable MAH, clean technical documentation, a QMS that actually meets Ministerial Ordinance NO. 169, and a post market surveillance plan ready from day one of commercialization.
The outcome is achievable. It just requires the right groundwork. Our Japan regulatory affairs services team supports manufacturers across classification strategy, MAH selection, PMDA submission preparation and QMS compliance. Start the conversation early. Reach out to us at info@artixio.com It makes everything that follows easier.
FAQs
Q1. How long does medical device registration take in Japan?
Class I takes 1 to 3 months. Class II certification takes 6 to 12 months. Class III and IV approval takes 12 to 24+ months. These are PMDA review timelines only. Preparation, documentation, translation and QMS assessment add considerable time before submission even begins.
Q2. What is the difference between an MAH and a D-MAH?
Same legal responsibility. Different setup. An MAH is a Japanese entity, typically a subsidiary or local company, that holds the product license directly under its own and infrastructure.
A D-MAH is a third party Japanese entity appointed by a foreign manufacturer to hold the license on their behalf. Same obligations, same accountability. Just not owned by the foreign manufacturer.
The risk with a D-MAH is capability. They are not all equal. Post market surveillance, adverse event reporting, recall management. If the D-MAH is not operationally strong in these areas, the foreign manufacturers carry the consequences.
Q3. Are clinical studies always required?
Not always. Depends on device class and available evidence. For many class II devices, published literature is sufficient. For Class III and IV, PMDA may require Japanese clinical data or strong justification for using foreign data.
Q4. What is the SAKIGAKE designation?
Japan’s expedited review pathway for genuinely innovative devices. Qualifying products receive priority PMDA consultation and accelerated timelines. Most manufacturers do not explore eligibility early enough to benefit from it.
Q5 What is the difference between certification and approval in Japan?
Certification applies to eligible Class II devices and is issued by Registered Certification Bodies. Approval applies to Class III and IV. It requires full PMDA technical review and takes significantly longer.

