Many regulatory delays in European Union happen not because of non-compliance, but because requirements are misunderstood early on. Different approval routes, shared responsibilities between authorities, and strict safety obligations make planning essential.
This article provides a practical view of EU pharmaceutical regulations and how they affect companies at each stage.
Pharmaceutical Regulations in European Union
Pharmaceutical regulations in the European Union (EU) are governed by a comprehensive framework aimed at ensuring the quality, safety, and efficacy of medicines by the central regulatory authority, European Medicines Agency (EMA). It is based in Amsterdam, Netherlands, and plays a critical role in the authorization and ongoing monitoring of medicines within the EU.
- The Regulatory framework for pharmaceutical products in the European Union (EU) is primarily governed by three main sets of regulations and directives:
- Regulation (EC) No 726/2004, a centralized procedure for the authorization of medicinal products in the EU. Under this procedure, certain types of medicines, such as biotechnology products, orphan medicinal products, and products for certain diseases, must be evaluated and authorized centrally by the European Medicines Agency (EMA).
- Decentralized Procedure and Mutual Recognition Procedure (Directive 2001/83/EC) – The mutual recognition procedure allows a pharmaceutical company that has obtained a national marketing authorization in one EU member state to apply for mutual recognition in other member states on meeting the necessary criteria.
- The decentralized procedure allows a company to apply for marketing authorization simultaneously in multiple EU member states, with one member state acting as the “Reference Member State” coordinating the evaluation, and other member states acting as “Concerned Member States” recognizing the reference member state’s decision.
Pharmaceutical Legislation Package:
Directive 2001/83/EC and Directive 2001/82/EC: These directives are part of the pharmaceutical legislation package and lay down the rules for the authorization, distribution, and pharmacovigilance of human and veterinary medicinal products, respectively.
Regulation (EC) No 726/2004: This regulation also forms a part of the pharmaceutical legislation package and provides the legal basis for the centralized procedure.
Application types of Pharma drugs in EU:
The three legal categories are discussed below, along with examples of drugs from each:
Biosimilars: A biosimilar is a biological drug that is very similar to another medicinal product that is already approved in the European Union (EU) but its marketing authorization may have expired. Before biosimilar medicines can be approved and marketed in the EU, the European Medicines Agency (EMA) must evaluate the majority of applications.
Generic and Hybrid applications: Generic/Hybrid medicines combine elements of both generic and originator medicines. They contain a previously authorized active substance (reference medicinal product) and may be developed for a new therapeutic indication or a new pharmaceutical form. Hybrid medicines must demonstrate that they are bioequivalent to the reference medicinal product in the same way as generic medicines.
Orphan Medicine are pharmaceutical products developed to treat rare diseases or conditions that affect a small population. In the European Union (EU), an orphan medicine is defined as a medicinal product intended for the diagnosis, prevention, or treatment of life-threatening or debilitating diseases that affect no more than five in 10,000 people.
Pediatric medicine refers to the branch of medicine focused on the medical care and treatment of infants, children, and adolescents, typically up to the age of 18 years. Pediatric medicine is a specialized field that addresses the unique healthcare needs of young patients, encompassing a wide range of medical conditions and preventive care.
Other types include Advanced therapy medicinal products (ATMPs) are medicines for human use that are based on genes, tissues, or cells. They present revolutionary new prospects for disease and injury therapy. And, for medicines intended for use outside the European Union, the European Medicines Agency (EMA), in collaboration with the World Health Organization (WHO), may provide scientific views on high-priority human medicines, including vaccines, intended for markets outside the European Union (EU).
Pharma Clinical trial requirements In EU:
Clinical trials for pharmaceutical drugs in the European Union (EU) are subject to strict regulatory requirements aimed at ensuring the safety, efficacy, and quality of the investigational products. These requirements apply to all stages of drug development, from initial testing in humans to post-marketing studies. The primary regulations governing clinical trials for pharmaceutical drugs in the EU are as follows:
Clinical Trials Regulation (CTR):
Regulation (EU) No 536/2014: The Clinical Trials Regulation, which was adopted in 2014, aims to harmonize and streamline the approval process for clinical trials across EU member states. EMA Management Board confirmed that the EU Portal and Database were fully functional. CTR has replaced the older directive 2001/20/EC and introduced several important changes to the clinical trial authorization process. [Directive 2001/20/EC, the earlier legal framework for conducting clinical trials in the EU until the Clinical Trials Regulation becomes fully applicable.]
Requirements for Clinical Trials in the EU:
Ethical Approval: All clinical trials conducted in the EU must receive approval from an independent ethics committee (IEC) or ethics review board (ERB) before they can commence. The IEC/ERB ensures that the trial design, participant recruitment, and informed consent process meet ethical standards.
Regulatory Approval: Clinical trials must also obtain approval from the competent national regulatory authorities in each EU member state where the trial is conducted. For multinational trials, a single application may be submitted to the “Reference Member State” for evaluation, and other member states may recognize the decision in a “Mutual Recognition” or “Decentralized” procedure.
Informed Consent: Participants must provide informed consent before participating in a clinical trial. The informed consent process ensures that participants are fully aware of the trial’s risks and potential benefits.
Good Clinical Practice (GCP): Clinical trials in the EU must be conducted in compliance with the principles of Good Clinical Practice, which sets international ethical and scientific standards for designing, conducting, and reporting clinical trials.
Pharmacovigilance: During the trial, adverse events and safety data must be collected and reported in adherence to pharmacovigilance regulations to ensure participant safety.
It is essential to note that regulations are subject to updates and changes, so researchers and sponsors should always refer to the latest guidance from the European Medicines Agency (EMA) and the competent national regulatory authorities for up-to-date information on clinical trial requirements in the EU.
Pharmaceuticals Labeling Requirements in EU
Product Information – Summary of Product Characteristics (SmPC)
Package Leaflet – Patient Information Leaflet (PIL)
Packaging and Labeling Requirements: The outer packaging of pharmaceutical products must include specific information, such as the product name, strength, pharmaceutical form, batch number, expiry date, storage conditions, and the name and address of the marketing authorization holder. Braille text on the packaging is required for certain medicines to enhance accessibility for visually impaired patients. Tamper-evident features must be incorporated into the packaging to ensure the integrity of the product.
Legal and Administrative Information: The marketing authorization number must be displayed on the packaging to indicate that the product is authorized for sale and use in the EU. The product must be labeled with its classification (e.g., prescription-only medicine, over-the-counter medicine) to inform appropriate dispensing practices.
Multilingual Labeling: The labeling, including the package leaflet, must be provided in the official language(s) of the EU member states where the medicine is marketed.
Pharma Products Registration Process In EU
Getting a pharmaceutical product approved in the EU doesn’t start with the EMA — it starts once your development work is done and you’re ready to defend what you’ve built.
After clinical trials, companies sit down and gather everything that tells the product’s story. How it’s made, what the studies showed, how safe it is, and where the risks lie. All of that material is pulled into one submission, commonly called the Marketing Authorization Application. There’s no shortcut here — regulators expect a clear, complete picture, not just results but the reasoning behind them.
Submission to the European Medicines Agency (EMA)
That application is then filed with the European Medicines Agency when the centralized route applies. From there, the EMA manages the review across EU countries, coordinating scientific questions and feedback. What’s reviewed isn’t just clinical data, but also manufacturing controls, labeling, safety plans, and how the product will be monitored once it’s on the market. The process is structured, yes — but success usually depends on how well the submission is prepared, not just what regulations say.
Documents that are required for the registration of a pharmaceutical drug in the EU are
Common Technical Document (CTD) –
- Module 1: Administrative and prescribing information.
- Module 2: Overall quality summary.
- Module 3: Quality data on the drug’s composition, manufacturing, and controls.
- Module 4: Nonclinical study reports (preclinical data).
- Module 5: Clinical study reports (clinical data)
- Summary of Product Characteristics (SmPC)
- Package Leaflet (Patient Information Leaflet – PIL)
- Risk Management Plan (RMP)
- Pharmacovigilance Plan
- After the application is sent, regulators first check one basic thing: is everything there or not?
They’re not judging the medicine yet. They just want to see if all the required forms, letters, and documents have been submitted properly. If something is missing, the process pauses until it’s fixed. - Once the file passes that check, experts review the medicine in detail. They look at how it was tested, how safe it appears to be, and whether it actually does what it claims to do. During this stage, questions often come back to the company, and clear answers matter.
- When that review is finished, a recommendation is made. The final approval itself comes from the European Commission. If the decision is positive, the medicine can be sold across the EU.
- That approval doesn’t last forever. It usually runs for five years. Before it ends, the company has to apply to keep it active. If they don’t, the approval simply expires.
EU Pharma Applicable charges:
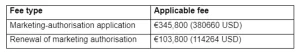
Pharma products approved in the US or other countries ease the registration process of pharma drugs in the EU. Approval from a regulatory authority such as USFDA may lead to Expedited Review. In the EU, the “Decentralized Procedure” or “Mutual Recognition Procedure” can be used to leverage data from a previous approval to support the EU application.
Post Marketing Surveillance
- Once a medicine is on the market, the work doesn’t stop there. Companies are expected to keep watching how it behaves in real use, not just how it performed during trials.
- If doctors or patients report side effects, those reports are collected and reviewed one by one. Some are minor. Some raise questions. Either way, they’re not ignored.
- All this safety information is shared with EU authorities on a regular basis so nothing sits in isolation. The idea is simple: if something starts changing, it should be noticed early.
- Over time, patterns can appear. A reaction might show up more often than expected, or only in certain patients. When that happens, the data is reviewed again to understand whether it’s coincidence or something that needs action.
- For some medicines, extra follow-up is needed after approval. That could mean additional studies or tighter controls to make sure risks stay manageable.
- If a safety concern is serious, information is circulated quickly across EU countries. Doctors are informed, guidance may be updated, and patients are kept in the loop where necessary.
- At the end of the day, everything comes back to one basic question: does the medicine still do more good than harm? That question keeps getting revisited as long as the product stays on the market.
Conclusion
For companies that require structured support, Artixio provides regulatory affairs consulting services in the European Union, assisting with strategy, submissions, and lifecycle management across EU and EEA markets. For enquiries related to EU regulatory or pharmacovigilance services, please write to info@artixio.com