
Pharmaceuticals in Japan are regulated by the PMD Act, published by the PMDA and MHLW. These are the two main regulatory authorities regulating pharmaceuticals in Japan. Reading this blog further will provide you with a brief overview of the regulatory requirements of pharmaceuticals in Japan.
Health Authority and Legal Representation In Japan:
All the pharmaceuticals in Japan are regulated by the PMDA and the MHLW. Below is a brief overview of both the regulatory bodies:
Ministry of Health, Labor and Welfare (MHLW):
- The MHLW is the Central Government regulatory body.
- It is responsible for amending policies, laws, and standards for governing pharmaceuticals, medical devices, and health policies in Japan.
- The MHLW has issued the PMD Act, which lays down the standards for pharmaceuticals to be regulated in Japan.
Pharmaceutical and Medical Device Agency (PMDA):
- The PMDA regulates under the MHLW.
- It is responsible for inspecting GMP, GVP, and GLP compliance.
- The PMDA provides approval for pharmaceuticals to be regulated in Japan.
- It monitors the post-marketing effects of pharma products and adverse effects.
- The PMDA is responsible for the pharmaceutical regulations from product development to market launch and post-marketing compliance.
Classification of Pharmaceuticals in Japan:
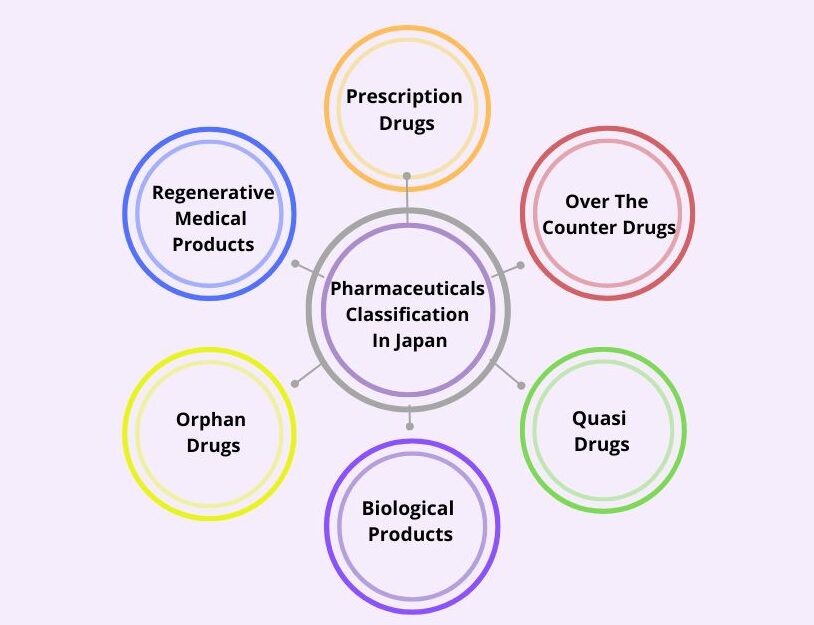
The pharmaceuticals are classified into various categories such as:
- Prescription Drugs: Pharmaceuticals sold under prescription.
- Over-the-counter Drugs: Pharmaceuticals sold without a prescription.
The OTC drugs are further classified as:
- Quasi Drugs: Products with mild therapeutic effects.
- Biological Products: Drugs derived from living organisms.
- Orphan Drugs: Medicines for rare diseases.
- Regenerative Drugs: Products developed from processed cells and tissues.
Pre-registration Requirements by PMDA:
Pharmaceutical companies engage in extensive studies, which encompass non-clinical research and clinical trials, preceding their pursuit of marketing authorization. These comprehensive studies generate crucial data that serves as the foundation for the subsequent registration application.
Documents Required by PMDA:
Following is the list of documents required for the pharmaceutical product approval in Japan:
- Quality Data
- Clinical Study Report
- Non-clinical Study Report
- Manufacturing Report
- Labeling Packaging Information
- Pharmacovigilance Plan
Registration of Pharmaceuticals In Japan:
The pharmaceutical in Japan undergoes a series of events before getting into the market. The steps are as follows: 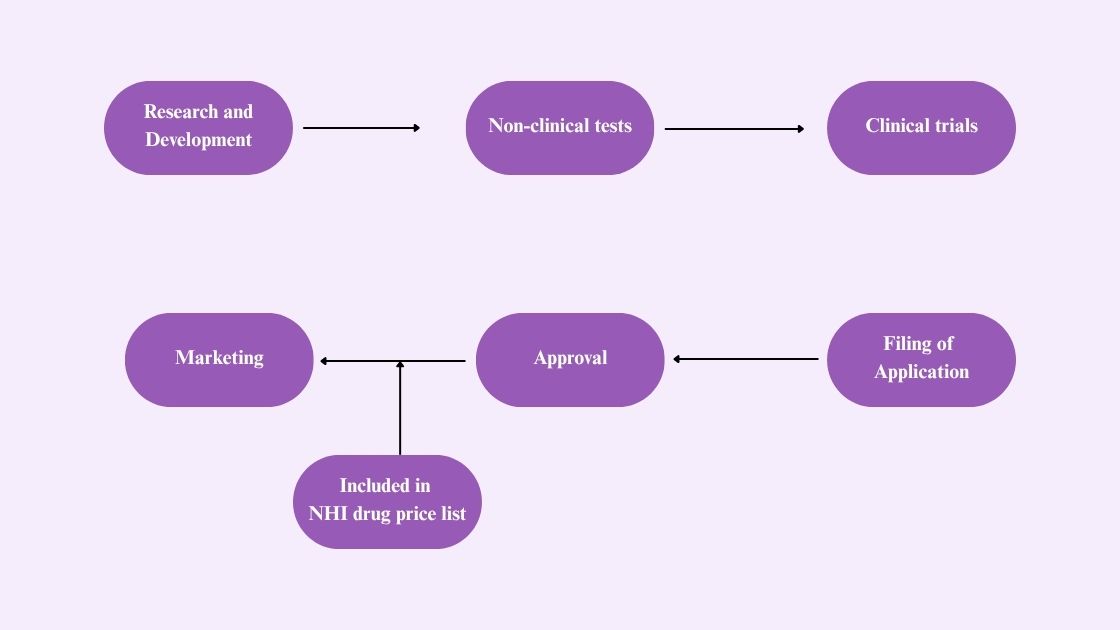 Here is the step by step Pharmaceutical Registration Process In Japan:
Here is the step by step Pharmaceutical Registration Process In Japan:
Research and Development:
- This is the first phase of the development of pharmaceutical drugs.
- By studying various aspects and after long years of testing and trial, the drug is formulated.
Non-Clinical Tests:
- In this step, various aspects of pharmacokinetic studies are performed and tested, such as absorption, distribution, toxicology, etc.
- These studies are conducted on animal models.
- The GLP guidelines are followed while performing non-clinical studies.
Clinical Tests:
- A Clinical Trial Notification (CTN) should be submitted to the PMDA for the initiation of the trial.
- All the required documents should be submitted along with the CTN.
- After approval from the PMDA, clinical trials can begin.
- The trials should be conducted in compliance with the Good Manufacturing Practice guidelines, keeping in mind the consent, safety, and health of the people engaging in the trial.
Filling of Application:
The application for the approval of pharmaceuticals in Japan is submitted in the form of an NDA through the PMDA’s Electronic Submission Gateway.
The NDA is submitted in a five Module format, which is:
- Module 1: Regional Information
- Module 2: CTD Summaries
- Module 3: Quality (Chemistry, Manufacturing, and Controls)
- Module 4: Non-clinical study reports
- Module 5: Clinical study reports
Following is the list of documents that should be submitted along with the NDA:
- Quality Data
- Clinical Study Report
- Non-clinical Study Report
- Manufacturing Report
- Labeling Packaging Information
- Pharmacovigilance Plan
This step of filling out the application is the official submission of all the documents, along with their approval.
Approval Of Pharmaceutical in Japan:
According to the PMDA’s review report, the MHLW approves the pharmaceutical to be regulated in Japan.
NHI Drug Price List:
- Once the drug is approved, it is enlisted in the NHI Drug Price List.
- In this step, the official price for selling the pharmaceutical in the Japanese market is decided.
Marketing:
- Once all the steps are completed, the pharmaceutical can be officially sold in pharmacies.
Regulatory Services Provided By PMDA:
Consultation:
The PMDA has a special consultation team, which provides consultation on various matters such as clinical consultation, R and D strategy consultation, and Regulatory affairs. The applicant, if he develops any difficulty or wants a consultation on any of the mentioned aspects before filling out the application, can consult the PMDA to utilize these services.
Regulatory Review:
This service is provided by the PMDA after filling out the application until the marketing phase. Here, the PMDA performs a pre-marketing review, and once the pharmaceutical is marketed, re-examination, re-evaluation, and use results evaluation are done when and as required.
GLP/GCP/GPSP Compliance Assessments:
GLP: Good Laboratory Practice or GLP is conducted during the non-clinical trials. This involves ensuring that standard laboratory practices are followed, and ethics are maintained while animal testing is conducted.
GCP: Good Clinical Practice or GCP is conducted by the PMDA during the clinical trial phase. The GCP sets standards to follow during the clinical trial. It involves various aspects like informed consent from the participants, data integrity, protocol compliance, etc.
GMP/QMS/GCTP Inspection: These inspections are carried out after the application is filled out until the marketing phase. These inspections aim to ensure that the manufacturing and quality standards are met and all regulatory requirements are fulfilled. Good Manufacturing Practice (GMP) ensures that the manufacturing standards are met, Quality Management System (QMS) ensures that the regulatory requirements are fulfilled and Good Gene, Cellular, and Tissue-based Products Practice (GCTP) ensures standards are followed while gene and living cells handling.
Standards Development:
The PMDA ensures that the standards are met during each step of the pharmaceutical lifecycle. Standard Development deals with the safety, quality, and efficacy criteria of the product. This Standards Development also provides safety measures and relief services for adverse health effects during the marketing and post-marketing phase.
PMDA Import And Distribution Requirements In Japan:
For pharmaceuticals to be imported and distributed, they should be approved by the PMDA. After its approval, it should comply with the importation and labeling standards.
Approval Timeline For Pharmaceuticals by PMDA:
The initial consultation takes a timespan of about 30 days whereas the subsequent consultation takes 14 days. Once the applicant submits the application, the PMDA reviews preclinical data, clinical study protocols, and other required documents.
Regarding associated fees, Japan’s PMDA charges various fees for different stages of the application process, including application fees, approval fees, and annual maintenance fees.
Foreign Testing and Applicability:
In order to ensure the quality, safety, and effectiveness of pharmaceutical products, they undergo a comprehensive testing process. This includes rigorous evaluations on raw materials, finished products, stability testing, and, if applicable, bioequivalence studies. These tests are crucial in maintaining the high standards of pharmaceutical products.
Approval in EU/US and Progressing Registration:
When it comes to obtaining approval for pharmaceutical products in Japan, it’s important to note that approval in the European Union (EU) or the United States does not automatically translate to approval in Japan. However, the data and findings obtained from research conducted in these regions can be utilized to bolster the registration application in Japan. Nonetheless, it’s crucial for companies to provide data that is specific to the Japanese population and adhere to the regulatory standards set by Japan. This ensures that the approval process in Japan remains meticulous and aligned with the unique needs and characteristics of its population.
Post-Marketing Activities:
Some of the post-marketing activities are as follows:
- Pharmacovigilance: The manufacturer must monitor the adverse drug reactions and report them immediately.
- Variations: Any variation in the product should be immediately reported to the PMDA.
- Renewal: The marketing authorization and product approval license should be approved before its expiration to keep your product in the Japanese market.
- Audits: Frequent audits are carried out by PMDA to ensure compliance by the manufacturer.
Conclusion:
The pharmaceuticals in Japan are regulated by the PMDA and MHLW. All the guidelines set by these authorities ensure a high-quality and efficient pharmaceutical. So, it can be concluded that establishing a pharmaceutical in Japan requires stringent regulatory compliance assuring the quality of the product.
For any regulatory assistance for your pharmaceutical in Japan, contact Artixio. We aim to provide you the ease to successfully navigate through your regulatory journey in Japan.
FAQs:
Can foreign manufacturers directly sell their pharmaceuticals in Japan?
No, foreign manufacturers intending to sell their pharmaceuticals in Japan need to appoint a Marketing Authorization Holder (MAH) in Japan and obtain a Foreign Manufacturer Accreditation from the PMDA.
What is National Health Insurance (NHI) Drug Price Listing?
The NHI Drug Price Listing is the official list where the drugs are listed once they’re approved. This is done for Japan’s universal health insurance system reimbursement.
What is the re-examination period for new drugs?
The re-examination period is the period in which the drugs, after approval, are examined to evaluate their safety and efficacy. This period is 8-10 years after drug approval.

