
Manufacturers are expected to have post-market surveillance as a component of their total quality management system. Post-market surveillance ensures the quality and safety of the medical device(s) over the entire lifecycle of the device. The regulation requires manufacturers to have actionable and data-ready PMS systems. A good PMS will both monitor for new risks, validate that the device is continuing to perform as well as intended, facilitate patient safety actions and ensure ongoing compliance with regulatory requirements.
Notified Bodies are placing greater emphasis on how manufacturers collect, review, and act on post-market data. As a result, companies need clear evidence that their PMS processes are operating effectively. In this article, we will look at the important requirements of PMS.
What is Post-Market Surveillance (PMS) Under EU MDR?
Upon the introduction of a medical device to the market, its performance should be monitored by manufacturers on an ongoing basis. Post-Market Surveillance is a process designed to verify the safety and effectiveness of the product.
An effective Post Market Surveillance process needs the collaboration of systems and procedures for creating an effective ‘picture’ on the performance of a particular medical device.
Why PMS is Important for Medical Device Manufacturers
PMS is important for manufacturers because compliance with the MDR proves that the device is safe and performing as per its intended use.
- Medical devices represent the backbone of the modern healthcare system.
- These activities are designed to generate information regarding the utilization of the device, to identify device design and/or usage problems, and accurately characterize the device behaviour in practice.
- The lack of harmonization within PMS results in an environment of increased adverse events involving MDs and overall mistrust in medical device diagnosis and treatment results.
PMS Requirements under EU MDR & IVDR
The PMS requirements are set out in Chapter VII, “Post-Market Surveillance, Vigilance and Market Surveillance,” specifically Article 83 of the EU MDR and Article 78 of the EU IVDR.
PMS Plan (PMSP)
A PMSP is part of the technical documentation required by the MDR. A PMSP includes a description of data collection and analysis activities. It also summarizes the methods used to collect information on serious incidents, field safety corrective actions, complaints, and user feedback.
As a part of the PMS system, the manufacturer must also establish procedure(s) to describe the activities of its PMSR, and PSUR.
A PMS plan serves as the framework for post-market monitoring activities. It shall contain the elements as per Annex III. Common elements include KPI tracking, case review, literature monitoring, trend analysis, risk management updates, and CAPA activities.
Annex III Requirements:
The manufacturer shall prove in a post market surveillance plan that it complies with the obligation referred to in Article 83
(a) The post market surveillance plan shall address the collection and utilization of available information, in particular:
- Information on serious incidents, including information from PSURs and FSCAs;
- Records relating to non-serious incidents and data on any undesirable side-effects;
- Information from trend reporting;
- Relevant specialist or technical literature, database and/or registers;
- Information, including feedback and complaints, provided by users, distributors, and importers; and
- Similar medical devices available in publicly available databases.
(b) The post market surveillance plan shall include at least:
- A systematic process for collecting and evaluating post-market data to monitor device performance and compare it with similar devices.
- Methods for assessing collected data, investigating complaints, and monitoring trends in incidents.
- Defined indicators and thresholds to support ongoing risk-benefit and risk management evaluations.
- Procedures for communicating with authorities, notified bodies, economic operators, and users.
- Processes for identifying issues, implementing corrective actions, and tracing affected devices when necessary.
The PMS plan should describe the methods used to analyze PMS data. Methods (e.g., qualitative, or quantitative, statistical methods) and processes for assessing the data are expected to be proportionate to the type of device. The methods selected are also dependent upon the type and quality of the collected raw data.
PMS Report (PMSR)
Manufacturers of Class I devices shall prepare a post-market surveillance report summarizing the results and conclusions of the analyses of the post-market surveillance data gathered as a result of the post-market surveillance plan referred to in Article 84, together with a rationale and description of any preventive and corrective actions taken.
The report shall be updated when necessary and made available to the competent authority upon request.
Periodic Safety Update Report (PSUR)
Manufacturers of Class IIa, Class IIb, and Class III devices must prepare a Periodic Safety Update Report (PSUR) for each device or device group, where applicable. The report should summarize PMS findings and conclusions and provide a rationale and description of any preventive or corrective actions taken. Throughout the lifetime of the device concerned, the PSUR shall set out:
- The conclusions of the benefit-risk determination.
- The main findings of the PMCF
- The volume of sales of the device and an estimated evaluation of the size and other characteristics of the population using the device, and where practicable, the usage frequency of the device
Submission frequency
- Manufacturers of class IIb and class III devices shall update the PSUR at least annually.
- Manufacturers of class IIa devices shall update the PSUR when necessary and at least every two years.
For class III and implantable devices, manufacturers shall submit PSURs through EUDAMED. For class IIa and class IIb devices, manufacturers shall make PSURs available to the notified body and, upon request, competent authorities through EUDAMED.
Trend Reporting Requirements
Methods and protocols to manage the events subject to trend reporting provided in Article 88 MDR and in Article 83 IVDR,
A reportable trend is any statistically significant increase in the frequency or severity of incidents that are not serious incidents, or in expected undesirable side effects. Such an increase could significantly affect the benefit-risk analysis and may lead to unacceptable risks to the health or safety of patients, users, or others.
In the context of trending, a “significant increase” should be determined by comparing the foreseeable frequency and/or severity of the incidents with the statistical methodology set out in the technical documentation.
Thresholds for frequency and severity of incidents or expected undesirable side effects/expected erroneous results should be considered when evaluating the criteria for Trend reporting.
Vigilance Reporting
Vigilance Reporting is the process for reporting, investigating, and managing serious incidents and corrective actions to maintain the safety and performance of medical devices after they are on the market.
- Manufacturers must report any Field Safety Corrective Action (FSCA) involving devices placed on the Union market. This requirement also applies to FSCAs carried out in third countries for devices that are legally available on the Union market.
- Information concerning serious incidents, including information from PSURs.
- Manufacturers shall report any serious incident involving devices made available on the Union market, except expected side-effects which are clearly documented in the product information and quantified in the technical documentation and are subject to trend reporting.
Reporting Timelines
Under the EU MDR and under the EU IVDR, the reporting timelines for serious incidents and Field Safety Corrective Actions (FSCAs) are:
| Event |
Reporting Timeline
|
| Serious public health threat |
Immediately, and no later than 2 days after awareness
|
| Death or unanticipated serious deterioration in health |
Immediately, and no later than 10 days after awareness
|
| Other serious incidents |
Immediately, and no later than 15 days after awareness
|
| Field Safety Corrective Action (FSCA) |
Report before implementing the FSCA, unless urgent action is required
|
PMS Requirements by Device Risk Classification
| Device Class | PMS Plan | PMSR | PSUR | PMCF |
| Class I | Post-market surveillance plan | Post-market surveillance report | Not applicable |
PMCF plan as per part B of Annex XIV, or a justification why a PMCF is not applicable
|
| Class IIa | Post-market surveillance plan | Not applicable | Periodic safety update report |
PMCF plan as per part B of Annex XIV, or a justification why a PMCF is not applicable
|
| Class IIb | Post-market surveillance plan | Not applicable | Periodic safety update report |
PMCF plan as per part B of Annex XIV, or a justification why a PMCF is not applicable
|
| Class III | Post-market surveillance plan | Not applicable | Periodic safety update report |
PMCF plan as per part B of Annex XIV, or a justification why a PMCF is not applicable
|
| Implantable Devices | Post-market surveillance plan | Not applicable | Periodic safety update report |
PMCF plan as per part B of Annex XIV, or a justification why a PMCF is not applicable
|
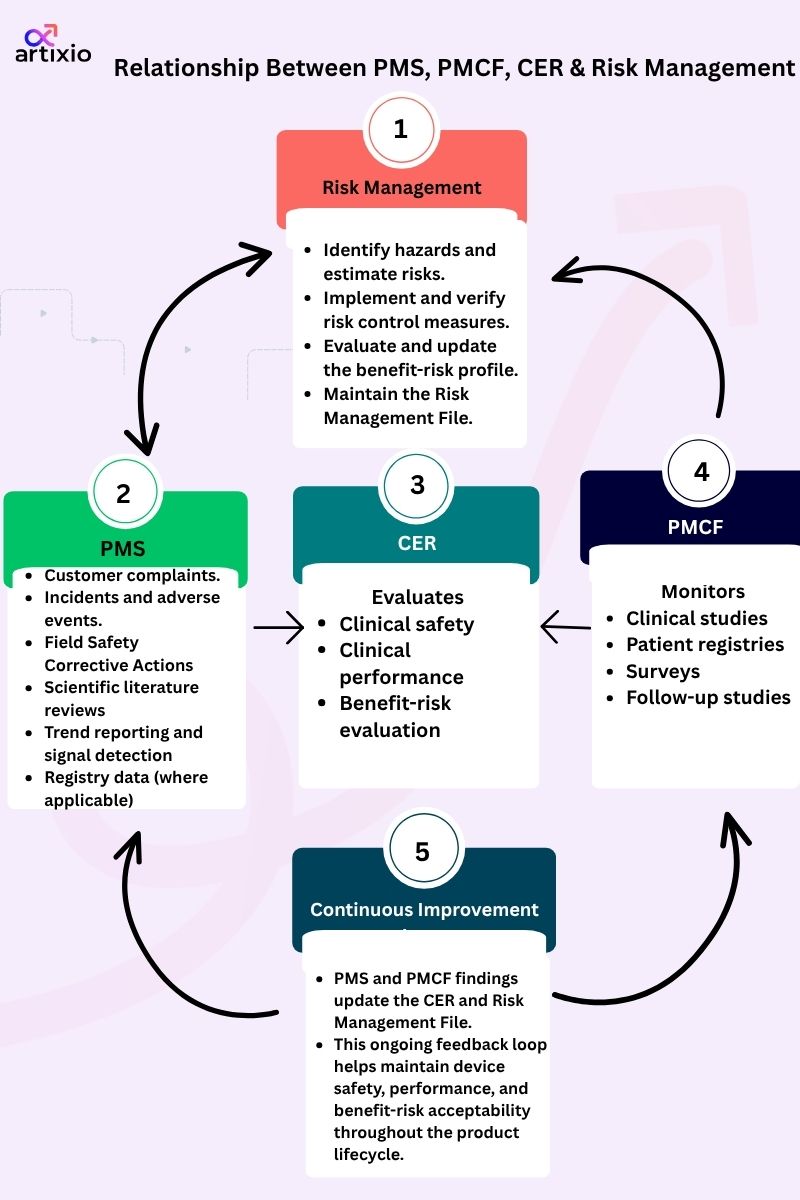
Relationship between PMS, PMCF, CER & Risk Management
PMS acts as the central post-market process. PMCF generates clinical data as part of PMS. The outputs from PMS and PMCF are used to update the Clinical Evaluation Report and the Risk Management File. When issues or trends are identified, CAPA activities may be initiated. All these activities are governed by the Quality Management System, which ensures that information flows between processes and is used to continuously improve the device, technical documentation, and regulatory compliance throughout the product lifecycle.
- Clinical data generated through PMCF is used to support the device benefit–risk ratio, support the claimed lifetime, and verify that the intended purpose statement is correct.
- PMCF is a continuous process that updates clinical evaluation.
- PMCF data generated under the directives may not be sufficient to demonstrate compliance with the EU-MDR, particularly for devices that can no longer claim equivalence.
- The relationship between risk management, clinical evaluation, PMS and PMCF is an ongoing process throughout the device’s life cycle that supports the safety, performance and benefit–risk profile of the device.
MDCG 2025-10 Guidance: Key Updates and Expectations
The publication of MDCG 2025-10 provides practical direction to implement PMS system. The document outlines how PMS activities fit within the broader quality system and describes the role of post-market information in identifying trends, reassessing risks, and supporting ongoing oversight of medical devices placed on the market.
Background of MDCG 2025-10:
This guidance is published as the manufacturers, notified bodies, and regulatory agencies were often unsure just how detailed, well-documented, and integrated their PMS activities needed to be. This document brings together the key PMS expectations. It explains how PMS should work from the moment a device is first developed.
This guidance focuses on risk-based PMS and lifecycle data analysis
Key Regulatory Clarifications:
- MDCG 2025-10 makes it clear that a Post-Market Surveillance (PMS) system is not just a formality. It is a continuous process that includes monitoring the functioning of a particular device, collecting information, analysing data collected, and reviewing the performance at various stages of the device life cycle.
- Monitor events before and after their occurrence and receive feedback to detect potential problems.
- Determine specific indicators and warning levels indicating the problem. This can detect when something is wrong, so action can be taken promptly.
- Make sure that PMS output becomes fully integrated into risk management, clinical evaluation, CAPA implementation, labeling, and technical documentation.
What has changed from previous guidance?
Before MDCG 2025-10, manufacturers relied on MDR Article 83, Annex III, ISO 13485, and notified body expectations when establishing PMS systems. While these requirements defined regulatory obligations, they provided limited practical guidance on integrating PMS planning, surveillance activities, reporting, and monitoring into a unified framework. Consequently, PMS implementation often differed from one manufacturer to another.
Expected Impact on Manufacturers
MDCG 2025-10 is likely to bring tighter oversight of PMS systems during notifier body evaluations and follow-up audits, meaning companies can expect more. Manufacturers won’t just need to show that PMS documentation is in place; they will also have to prove they are consistently gathering, reviewing, and acting on that data to inform regulatory decisions.
Manufacturers are supposed to
- Look at the current PMS plans and procedures and make sure they line up with the updated guidance.
- Make it easier to track how PMS findings connect to updates in risk management.
- Make sure the PMS reports directly tie into how patient care or clinical evaluations are updated.
- Ensure to keep clear justifications, If PMCF activities are not carried out.
Common PMS Non-Conformities Identified by Notified Bodies
The most common non-conformities that Notified Bodies frequently identify in PMS systems are:
- Companies often don’t fully embed post-market surveillance into their quality management systems, or they lack a strong, consistent approach to it.
- Many manufacturers adopt approaches which are incomplete, unclear, and not device specific.
- Rather than a more proactive methodology that looks at data in real time, manufacturers tend to be reactive when dealing with incidents.
- It is common for manufacturers to misinterpret trends, and there is not always a strong connection between PMS outputs and other key processes like risk management, clinical evaluations, PMCF, or CAPA.
- Notified Bodies often point out key gaps, such as weak oversight, inconsistent trend reporting, incomplete records of PMS activities, and missing clear metrics or thresholds.
How to Implement an Effective PMS System under EU MDR
- The manufacturer should have knowledge about Post-Market Surveillance (PMS) and how the process takes place throughout the entire lifecycle of the device.
- To develop an efficient post-market surveillance system, it is necessary to determine roles, methods for gathering and assessing data, and the sources of information that are to be analyzed.
- Furthermore, Post-Market Clinical Follow-up (PMCF) should be performed for collecting additional clinical data and ensuring the safety and performance of the device.
- Continuous analysis of complaints and adverse effects allows manufacturers to address new issues and act upon them in a proper manner.
PMS Documentation Checklist for EU MDR Compliance
To show compliance with the EU MDR, manufacturers must keep a full and well-organised collection of PMS documents. Essential documentation comprises a PMS Plan, PMS Report, or Periodic Safety Update Report (PSUR), a Post-Market Clinical Follow-Up (PMCF) Plan and report. Maintaining these documents enables manufacturers to demonstrate that device safety and performance are consistently tracked throughout the product’s lifecycle.
How Regulatory Consultants Support PMS Compliance
Regulatory consultants assist manufacturers in setting up and sustaining compliant PMS systems in accordance with EU MDR standards. Their support may include developing PMS Plans, preparing PMS Reports and PSURs, conducting PMCF activities, managing complaint and vigilance processes, and maintaining regulatory documentation. Consultants also support EU MDR gap assessments, internal audits, risk management tasks, and communications with Notified Bodies, enabling manufacturers to detect compliance issues early and sustain an effective post-market surveillance system across the device’s entire lifecycle.
Conclusion
Under the EU’s MDR and IVDR regulations, Post-Market Surveillance (PMS) is a hands-on effort that companies must keep up with. It is proactive, not reactive; it is fully woven into the manufacturer’s Quality Management System and works together with key processes like post-market patient monitoring, clinical evaluations, risk management, and corrective actions. PMS helps manufacturers monitor real-world data in a structured manner. This enables timely updates to documentation and implementation of corrective actions when needed.
Need support with PMS compliance? Our EU regulatory experts can assist with the development of PMS Plans, PMS Reports, PSURs, PMCF documentation and overall EU MDR/IVDR post-market surveillance requirements. Contact us at info@artixio.com. To discuss how we can support your regulatory compliance needs.

