
Ask any regulatory consultant which medical market to focus on first in Europe. The answer is almost always Germany. And the reason is simple. It is the biggest market in Europe, and cracking it sets the template for everything else.
Germany leads Europe by market size as well as by regulatory complexity. Every device used by patients in Germany goes through a rigorous regulatory system. Manufacturers selling these devices in Germany must comply with the European Union Medical Device Regulation (EU) 2017/745 (MDR) and the national obligations.
Artixio has noticed a consistent pattern. The companies that move faster are the ones who understood this regulatory structure and obligations before they started. And this article provides exactly everything you need to know about Germany Medical Device Regulatory Requirements.
Regulatory Authorities for Medical Devices in Germany
Germany medical device regulations are enforced by a combination of European Union authorities and German national component authorities. Each agency handles a completely different aspect of the approval process. Their shared goal is making sure that the medical devices marketed in Germany are safe and perfectly aligned with the mandatory requirements.
1. European Commission – Sets the Rules
The European Commission creates the rules that all medical device manufacturers must follow across the EU. The regulatory requirements for medical devices is established through:
- Medical Device Regulation (EU) 2017/745 (MDR)
- In Vitro Diagnostic Medical Device Regulation (EU) 2017/746 (IVDR)
2. BfArM – Germany’s Main Medical Device Authority
The Federal Institute for Drugs and Medical Devices (BfArM) operates under the German Federal Ministry of Health. It is Germany’s main medical device regulatory body and serves as the key contact point for manufacturers and regulatory activities.
It is responsible for monitoring devices on the market, adverse event reporting, approving clinical studies, checking regulatory compliance and coordination with EU regulatory systems.
BfArM’s Medical Device Information and Database System (DMIDS) portal is used by companies for clinical investigations and certain regulatory submissions.
3. State level Authorities – Monitor the market
Germany has a federal system. The regulatory responsibilities in Germany are shared between the national government and individual German states (Länder). The state authorities ensure that medical devices continue to comply with regulations after they are sold in Germany.
4. Notified Bodies – Assess Devices Before Market Entry
Notified Bodies are chosen under the EU Medical Device Regulation (MDR) as an independent organization. Assessment of higher risk medical devices compliance with regulatory requirements are done by these bodies. They also issue the certification needed to obtain CE (Conformité Européenne) marking.
Germany Medical Device Classification
Under the EU Medical Device Regulation (MDR) 2017/745, medical devices are grouped into four risk-based classes. Class I, Class IIa, Class IIb and Class III.
| Class | Risk | Medical Devices |
| Class I | Low |
Non-sterile bandages, Examination gloves, Manual wheelchairs
|
| Class IIa | Medium |
Dental filling materials, Ultrasound equipment, Hearing aids
|
| Class IIb | Medium to High |
Ventilators, Infusion pumps, Orthopedic implants
|
| Class III | Highest |
Heart valves, Coronary stents, Implantable pacemakers
|
Class I medical devices have additional classifications based on the specific characteristics.
Class Is is a low risk medical device that is sterile (surgical gloves). Class Im is a low risk device that measures something (medical thermometer). Class Ir is a low risk reusable surgical instrument (surgical scissors).
Most classification challenges happen in Class IIa and Class IIb categories. Medical software and substance based devices often are in the grey area which can cause disagreements between manufacturer and a Notified Body to select the correct class. The disagreement issue is then reviewed by the Competent Authority in the relevant EU member state. Other regulators may also be consulted if needed. The Medical Device Coordination Group (MDCG) and the European Commission are notified in certain cases.
Artixio has observed that classification issues are the most common reason medical device market entry gets delayed. So a proper classification assessment can prevent months of delays and unnecessary cost.
EC REP Requirements for Medical Devices
Appointing an EU-based Authorized Representative (EC REP) is mandatory for manufacturers based outside the EU. EC REP serves as the main point of contact for BfArM and other national authorities.
What they do:
- Keep technical documents and the Declaration of Conformity available
- Work with regulatory authorities when needed
- Help with safety issues and incidence reporting
- Support compliance with MDR requirements
The EC REP’s name and address must be shown on the device label.
Medical Device Regulatory Guidelines in Germany
There are three layers that are applied. EU regulation, German national law and German administrative rules.
Key Regulations:
| Regulations | Purpose |
| MDR (EU 2017/745) |
Main EU law for medical devices. Sets requirements for safety, performance, CE marking and market access.
|
| MPDG |
Germany’s main law for enforcing MDR and IVDR
|
| MPDV |
Provides detailed administrative and procedural rules
|
| MPBetreibV |
Governs the safe operation and maintenance of medical devices in healthcare facilities
|
Medical Device Registration Process in Germany
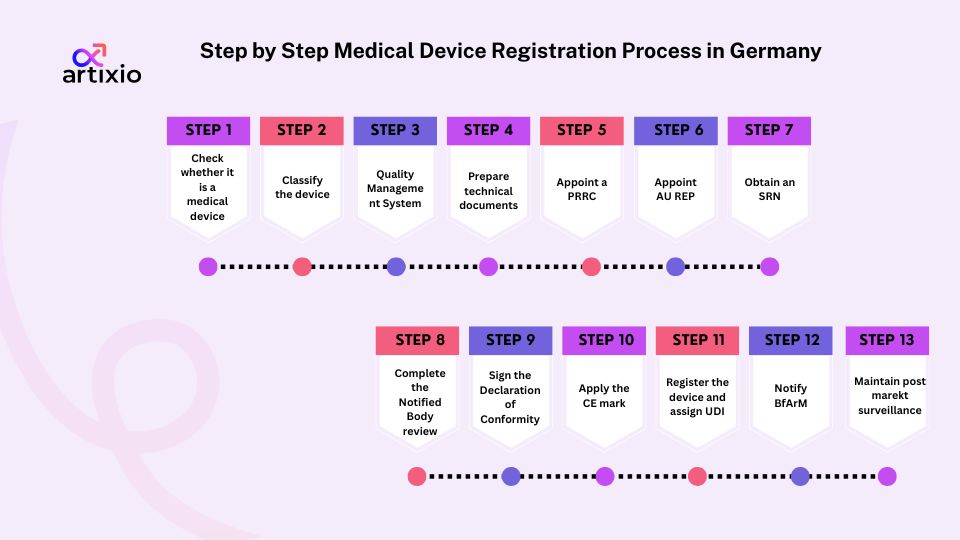
Here is the step by step approval process.
- Step 1: Check whether it is a medical device
Understanding the Germany medical device registration process starts with knowing what qualifies as a medical device in the first place. Software and combination products often need a closer look. - Step 2: Classify the device
Then comes classification. The MDR groups devices into different risk classes. Classifying the device into the wrong category can create hassle later. - Step 3: Set up a Quality Management System (QMS)
Before moving ahead, put QMS in place. It should cover quality, safety, risk management and regulatory requirements. - Step 4: Prepare the required technical documents
Collect the evidence of safety and performance in an organized way. - Step 5: Appoint a PRRC
A qualified Person Responsible for Regulatory Compliance (PRRC) must be designated MDR expects someone to oversee compliance activities - Step 6: Appoint an EU Authorized Representative (if needed)
An Authorized Representative is required for manufacturers based outside the EU. They act as the main contact within the EU. - Step 7: Obtain an SRN
Registration in the European Database on Medical Devices (EUDAMED) leads to a Single Registration Number (SRN). Think of it as your official regulatory identifier. - Step 8: Complete the Notified Body review (if applicable)
Higher risk devices must undergo assessment by a Notified Body. Lower risk Class may not need this step - Step 9: Sign the Declaration of Conformity
A Declaration of Conformity is prepared and signed once compliance is demonstrated . This formally confirms MDR compliance. - Step 10: Apply the CE mark
Now the device can receive its CE mark. That’s the green light for placing it on the EU market. - Step 11: Register the device and assign a UDI
Device details are entered into EUDAMED and a Unique Device identifier (UDI) is assigned. - Step 12: Notify BfArM
Before marketing the device in Germany, a notification must be submitted to BfArM. This applies to all device classes. - Step 13: Maintain post market surveillance
Monitoring and complaint handling remain part of the process throughout the product’s lifecycle.
Required Documents for Medical Device Registration
Getting a device registered in Germany means building a document set that covers several distinct areas. The heavier the device’s risk class, the heavier the file.
Starting with basics. Manufacturer information, device registration details, a Declaration of Conformity, and an Authorized Representative agreement for manufacturers from outside the EU. These are administrative foundations.
Then comes the technical documentation. Device description, intended use statement, design and manufacturing details, risk management file is all that needed. Depending on the device, expect to provide a Clinical Evaluation Report, performance testing data and verification and validation records. For higher classes, this section alone can take months to build properly.
In quality compliance documentation, an Iso 13485 certificate is standard. Supporting QMS procedures, CAPA records and supplier control documentation should be in order.
For post market obligations, a PMS plan is typically required from the start. A PMCF plan may be needed depending on the device, along with documented vigilance procedures.
Not every device needs every document. Class and intended use determine what is mandatory and what is conditional.
Germany QMS Requirements for Medical Devices
MDR requires QMS to be appropriate for the device’s risk level. A strong QMS is essential for Germany MDR compliance.
ISO 13485 Compliance
Most manufacturers use ISO 13485 as the guiding standard for their QMS. It covers:
- Design controls
- Supplier management
- Production control
- Complaint handling
- CAPA
- Post market surveillance
Certification and Ongoing Maintenance
An ISO 13485 certificate remains valid for 3 years. During those 3 years, annual surveillance audits are carried out. Meanwhile, CAPA records, internal audits and management reviews need regular attention.
Medical Device Labelling Requirements in Germany
Every device entering the German market carries mandatory information. Medical device labeling must comply with MDR requirements.
Mandatory Label Information
- Device name
- Manufacturer name and address
- Authorized Representative details (if applicable)
- CE marking
- UDI information
- Batch or serial number
- Intended purpose
- Warnings and precautions
- Sterility information (where applicable)
Language Requirements
Information intended for users in Germany generally must be provided in German, particularly instructions for use and safety related information. This is unless expectations are applied under national regulations. Translation quality matters too. For Class III devices especially, using an ISO 13485 certified translation provider is strongly recommended to ensure accuracy and patient safety.
Registration Timelines for Medical Devices in Germany
Actual timeline depends upon device classification and Notified Body involvement
- Class I – 1 to 3 months
- Class Is/Im/Ir – 3 to 6 months
- Class IIa – 6 to 12 months
- Class IIb – 9 to 18 months
Medical Device Registration Costs in Germany
Several factors that influence the overall cost are:
- Device classification
- Notified Body fees
- Clinical evaluation requirements
- Product testing and validation
- Quality Management System (QMS) certification
- Regulatory consulting or compliance support
There is no fixed registration fee that applies to every device. For lower risk devices, expenses may be relatively modest. Higher risk products, especially Class III devices involve much greater costs due to more extensive reviews and clinical evidence requirements.
Import Requirements for Medical Devices in Germany
Before anything reaches the market, the importer checks three things. Valid CE mark on the device. Manufacturer and EC REP obligations fulfilled. And lastly the correct labelling and instructions for use in German language where required.
Registration in EUDAMED is mandatory. German importers are required to register in the European database for medical devices, with further guidance available through the EUDAMED information Centre.
Documentation doesn’t stop at market entry either. Copies of the Declaration of Conformity need to be kept. Technical documentation must be available on request.
Germany medical device import requirements make an EU importer a legal requirement for manufacturers based outside the EU. And under the MDR, that importer carries real regulatory weight. Manufacturers and imposters must comply with MDR importer obligations.
Post-Market Surveillance (PMS) Requirements in Germany
To meet PMS Requirements, manufacturers must:
- Collect post-market data
- Analyze complaints and incidents
- Maintain PMS reports
- Update risk management files
- Implement corrective actions when necessary
- Vigilance Reporting
Whenever safety concerns arise, corrective actions may be necessary. In some cases, product recall and safety notices may be implemented. Under MDR vigilance requirements, serious incidents and Field Safety Corrective Actions (FSCAs) must be reported to the relevant authorities within the applicable timelines. A regular safety reporting is required for higher class devices. These reports are known as Periodic Safety Update Reports (PSURs). They provide an overview of the device’s safety and performance data.
Where additional clinical evidence is needed, Post-Market Clinical Follow-up (PMCF) activities should be carried out. The goal is to confirm that the device remains safe and compliant throughout its lifecycle.
PMS is an ongoing process. It helps manufacturers identify issues early and maintain compliance with MDR requirements in Germany.
Conclusion
The German medical device market is worth entering. It is also unforgiving for underprepared applications. CE marking, BfArM notification, German language documentation, active post market surveillance and MPDG compliance are all mandatory German medical device regulatory requirements. The manufacturers who navigate this well are the ones who started preparing early and chose their Notified Body and EC REP carefully. And that preparation is exactly where Artixio comes in.
Whether you are mapping your first entry into Germany or untangling a compliance gap mid-process, Artixio’s regulatory team works to get your devices market ready. Get in touch with Artixio at info@artixio.com to map a practical and specifically built Germany regulatory pathway for your device.

