
South Korea has built one of the most demanding medical device approval systems in Asia. The Ministry of Food and Drug Safety (MFDS) is the main regulatory body and its pre-market approval process operates very differently from the FDA or EU.
The cost of getting the registration process wrong is an expensive mistake. MFDS compliance matters from day one. Getting it right and it will reduce the risk of recalls and quality failures, build trust with healthcare professionals and keep the door open for sustainable business growth in the market.
This article gives detailed information on the process of registration of medical devices in South Korea.
Regulatory Authority for Medical Devices in South Korea
Manufacturers planning to commercialize medical devices in South Korea must follow a structured approval framework which is regulated by the Ministry of Food and Drug Safety (MFDS).
The regulatory framework sits under the Medical Device Act supported by the Enforcement Decree and Enforcement Rule. Together these documents define which devices need approval versus simple notification, what documentation is required, how manufacturing compliance is assessed and what obligations stay active after a product is on market.
Two other bodies matter beyond MFDS itself. First is the National Institute of Medical Device Safety Information (NIDS) which provides technical guidance and handles international harmonization matters. Then there are designated testing and certification bodies that carry out laboratory assessments required during the approval process.
MFDS Medical Device Classification
In South Korea, device classification determines everything including registration pathway, documentation requirements, timeline and cost.
Korean classification codes are device-specific rather than purely category-based. Manufacturers should verify their device’s exact classification through the MFDS Medical Device Information & Approval Portal before committing to any documentation work.
| CLASS | RISK LEVEL | EXAMPLES | PATHWAY | KGMP REQUIRED |
| Class I | Low | Bandages, tongue depressors, basic surgical instruments | Notification | No (voluntary) |
| Class II | Low-medium | Ultrasound equipment, powered wheelchairs, syringes | Notification with documentation | Yes |
| Class III | Medium-high | Orthopedic implants, dialysis machines, ventilators | Full pre-market approval | Yes |
| Class IV | High | Active implantable, cardiac stents, HIV diagnostics | Full pre-market approval = clinical data | Yes (mandatory audit) |
MFDS medical device registration process in South Korea includes both notification and approval pathways, depending on the level of product risk. While the registration pathway varies based on device classification, all manufactures must address following regulatory requirements.
- Korean License Holder (KLH)
- KGMP Certification
- Technical Documentation
- Performance & Safety Testing
- Clinical Evaluation (where applicable)
- Korean Labeling & IFU
- Post- Market Surveillance Compliance
These requirements vary based on device classification and registration pathway and are discussed in detail in the further sections.
Medical Device Registration Process in South Korea
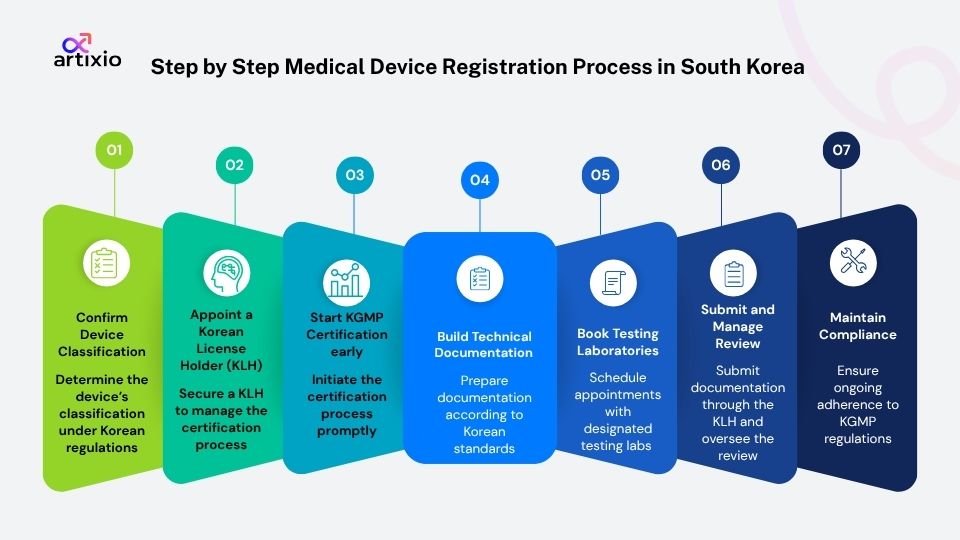
Here is the detailed step by step process of medical devices registration in South Korea for fast approval:
Step 1: Confirm Device Classification
MFDS assigns device-specific codes based on intended use and the same product can land in a completely different class. A wrong classification at the start means redoing work you have already paid for.
Step 2: Appoint a Korean License Holder (KLH)
You cannot hold a Korean marketing license as a foreign company. A Korean legal entity must hold it for you and take on real legal responsibility for your device in the market. Choosing the right one early matters more than most companies expect.
Step 3: Start KGMP Certification early
KGMP is the single most common cause of registration delays for foreign manufacturers. If you hold a current ISO 13485 certification, MFDS may accept an equivalence review instead of a full inspection, but do not assume that. Confirm it for your specific device class with your KLH before counting on it.
Step 4: Build Technical Documentation to Korean Standards
The content is similar to what you would put in an EU technical file but every section needs certified Korean translation and MFDS has its own formatting expectations. There are two sections that most commonly cause delays. First is the Essential Principle of Safety and Performance checklist, where incomplete justifications for non applicable items consistently draw MFDS queries. Second is the risk management file where submissions based on the outdated ISO 14971:2007 standard rather than the current 2019 version get flagged during the review.
Step 5: Booking MFDS Designated Testing Laboratories Early
MFDS only accepts test reports from its designated laboratory list. It does not matter if your existing test reports used identical methodology. If the lab is not on the list, the reports do not count. Book early because these labs have limited capacity and their lead times regularly become the critical path on registration timelines.
Step 6: Submit Through Your KLH and Manage the Review
Your KLH handles the MFDS submission. Class I and II devices typically clear in one to three months. Class III and IV go through full technical review which runs anywhere from 6 months to over eighteen months for complex devices. MFDS will send queries during review and the response windows are quite strict so stay in regular contact with your KLH .Every Foreign manufacturer seeking to sell a medical device in South Korea must appoint a Korea License Holder (KLH).
Step 7: Maintain Compliance After Approval
License needs renewing every five to six years. KGMP certificates expire every three years. Serious adverse events must be reported within 15 days, non serious within 30 days. Companies that go quiet after approval are the ones that get caught out by renewal deadlines or PMS obligations they forgot were coming.
Required Documents for MFDS Registration
| DOCUMENT | CLASS I | CLASS II | CLASS III | CLASS IV |
| KLH application form | ✓ | ✓ | ✓ | ✓ |
| Technical documentation | Basic | Full | Full | Full |
| KGMP certification | _ | ✓ | ✓ | ✓ |
| Performance test reports | _ | ✓ | ✓ | ✓ |
| Clinical Evaluation | _ | Sometimes | ✓ | ✓ |
| Risk management file | _ | ✓ | ✓ | ✓ |
| Korean labeling + IFU | ✓ | ✓ | ✓ | ✓ |
| Free Sale Certification | Sometimes | Sometimes | Often | Often |
Korea License Holder (KLH) Requirements
Korea License Holder (KLH) cannot hold a medical device marketing License in South Korea as a foreign company, it must be held by a Korean legal entity acting as your Korean License Holder. Your KLH is legally accountable for the device in Korea. KLH is the contact point between the manufacturer and MFDS.
KLH responsibilities may include adverse event reporting, product recalls, labeling compliance, renewal management, and maintaining regulatory documentation within South Korea. Choosing a reliable KLH is especially important for companies commercializing higher-risk or technologically complex products, including AI-driven medical devices and SaMD solutions.
Foreign manufacturers who need an experienced KLH or regulatory partner can explore Artixio’s South Korea MFDS compliance services.
KGMP Certification requirements
Korea Good Manufacturing Practice (KGMP) is South Korea’s quality management system standard for medical device manufacturing. KGMP is closely aligned with ISO 13485 but enforced through a distinct Korean certification process.
For domestic Korean manufacturers, KGMP inspections are conducted by MFDS or a designated inspection body at the manufacturing site. For overseas manufacturers, MFDS may accept an equivalence review based on existing ISO 13485 certificates or conduct an on-site overseas inspection, particularly for Class IV devices or where questions arise about manufacturing.
One thing manufacturers frequently underestimate is the Korean language documentation requirement. The KGMP assessment process may require certain documents in Korean, and quality system records will need to be translatable on request. Having a KLH or regulatory partner who can handle this interference is essential.
KGMP certificates are time limited and require renewal. If your certificate lapses, your marketing license is at risk.
Technical Documentation, Labeling and IFU Requirements for Medical Device
Technical documentation is the core of any MFDS registration process. For Class II, III and IV it covers roughly the same ground as EU technical file or FDA design file but with some Korea-specific additions and formatting conventions.
Technical documentation must cover the full design, manufacturing, safety and performance lifecycle of the device. Every section of a technical file that is not originally drafted in Korean must be accompanied by a certified Korean translation.
| TECHNICAL DOCUMENTATION SECTION | REQUIRED FOR CLASS | KEY STANDARD |
COMMON MISTAKES
|
| Device Description and Intended Use | All classes | MFDS submission template |
Vague statements that do not match Korean classification code
|
| Design and manufacturing information | Class II – IV | KGMP / ISO 13485 |
Mismatched between KGMP certificate scope and actual manufacturing process described
|
| Essential Principles of Safety and Performance | Class II – IV | MFDS EPSP guidance |
Incompletely addressed principles and missing justification for non-applicable items
|
| Risk management file | Class II – IV | ISO 14971:2019 |
Outdated ISO 14971:2007 format and risks not linked to test evidence
|
| Performance and safety test reports | Class II – IV | Korean KS / IEC / ISO standards |
Reports from non- MFDS designated labs and expired test certificates
|
| Clinical evaluation summary | Class III – IV (Sometimes Class II) |
MFDS clinical guidance |
Literature search too narrow and equivalence claims not adequate substantiated
|
| Post-market surveillance plan | Class II – IV | Medical Device Act PMS rules |
Generic plans not tailored to the specific device and risk profile
|
| Korean labelling and Instructions for use (IFU) | All classes | MFDS labeling regulations |
Missing mandatory labelling elements and non compliant font or symbol usage.
|
Clinical evaluation requirements in South Korea
MFDS requires a clinical evaluation report (CER) for class III and class IV medical devices. The CER must demonstrate device safety and clinical performance through a systematic review of clinical literature and clinical investigation data. Equivalence claims in which the manufacturer relies on clinical data from similar devices must be subjected to a rigorous demonstration of technical, biological and clinical equivalence evidence. MFDS also accepts clinical data from other regulatory bodies (FDA, CE) as supporting evidence when appropriately presented.
| CER REQUIREMENT | CLASS I | CLASS II | CLASS III | CLASS IV |
| Clinical evaluation report | Not required | Sometimes | Required | Required |
| Systematic evaluation review | _ | Sometimes | Required | Required |
| Clinical investigation data | _ | Rare | Sometimes required | Often required |
| Equivalence approach accepted | _ | Yes | Yes (with strong justification) | Limited |
| Post-market clinical follow-up plan | _ | Recommended | Required | Required |
| Foreign clinical data accepted | _ | Yes | Yes (as supporting evidence) |
Yes (but Korean data preferred for novel devices)
|
Testing and Safety Requirements for MFDS Registration
Performance and safety testing is a mandatory component of MFDS registration for Class II, III, and IV devices. MFDS requires that test reports come from MFDS designated testing laboratories. Reports labs that aren’t on MFDS’s list may not be accepted for primary submission data, even if the testing methodology and standards applied were identical. This requirement has practical implications for timeline planning, since these laboratories have finite capacity and can only accept bookings when slots are available.
Common tests include electrical safety (IEC 60601), biocompatibility (ISO 10993), EMC testing, sterility validation and device specific performance tests.
Registration Timelines for Medical Device in South Korea
The registration time for medical devices in South Korea will depend on the medical device classification and regulatory pathway. Medical devices with low risk typically go through a faster review process because there is no active MFDS technical review. High risk are subject to greater technical and clinical assessments as for them full technical review is required.
| Device Class | Total Time |
| Class I and II | 1-3 months |
| Class III and IV | 6-18 + months |
MFDS Registration Costs for Medical Devices
The total costs for MFDS registration will vary device type, testing requirements, technical documentation, KGMP certification and KLH arrangements. In addition to these, manufacturers should also consider expenses related to document translation and ongoing compliance. As higher risk devices are subject to more extensive regulatory requirements. Registration costs generally increase with device classification.
| Device Class | Key Cost Driver |
| Class I |
Simplified registration and limited review.
|
| Class II |
Technical documentation and testing requirements
|
| Class III |
Detailed technical review, testing and KGMP compliance
|
| Class IV |
Clinical evidence and comprehensive review
|
Post-market Surveillance Requirements
Once a device is on the Korean market, post-market surveillance obligations kick in and remain in place for as long as the device is marketed. The KLH carries primary responsibility for managing PMS obligations in Korea, but the manufacturer must ensure that safety information flows from their end to the KLH in a timely and structured way because if MFDS reporting deadlines are missed, it’s a serious compliance failure.
| PMS OBLIGATION | WHO IS RESPONSIBLE | TIMEFRAME |
| Serious adverse event reporting | KLH (informed by manufacturer) | Within 15 days |
| Non serious adverse event reporting | KLH | Within 30 days |
| Periodic safety report | KLH + manufacturer |
Per MFDS schedule
|
| Recall / field safety action | KLH (MFDS oversight) |
Immediately on decision
|
| Complaint records | Manufacturer + KLH |
Maintained continuously
|
South Korea Medical Device Import Requirements
Importing medical devices into South Korea requires more than a valid MFDS marketing license. The physical importation process has its own requirements that news to be managed in coordination with the KLH.
Devices must carry Korean language labeling before they enter distribution. Whether this labeling is applied by the overseas manufacturer before shipment, or by the KLH after the import in a Good Manufacturing Practice (GMP) controlled environment, needs to be agreed and documented as part of the product’s quality management system. Relabeling in Korea is permitted but must be done under controlled conditions and documented appropriately.
From a customs standpoint, medical devices are classified under specific HS code and may be subject to import duties. But South Korea’s Free Trade Agreement (FTA) with the EU,US, and several other major trading partners means that import duty rates for many medical devices are reduced or eliminated.
For devices that have received MFDS approval, a copy of the marketing license and relevant product documentation should accompany or be available for each import shipment to facilitate customs clearance. The KLH coordinates the import process and maintains the necessary records.
Conclusion
Korea is demanding on paper and in practice. But manufacturers who go in prepared with classification confirmed, a reliable KLH in place, KGMP addressed early and documentation built for MFDS specifically can consistently get through the process faster and with few surprises.
If you are not sure where your device sits in Korea’s medical device classification system or whether your existing ISO 13485 certificate will satisfy KGMP requirements, those are good questions to bring to us early. Learn more about our South Korea regulatory affairs services or reach out to info@artixio.com.

