
The pharmaceutical market in Australia is projected to reach USD 9.78 billion by the end of the year 2023 with the largest market share of oncology drugs which is projected to reach USD 1.80 billion.
The revenue in the pharmaceuticals market is growing with an annual rate of 6.54% from 2023 to 2028 and the projected market volume by 2028 is US$13.43bn.
Pharmaceutical Regulatory Authority in Australia
Pharmaceutical drug regulations in Australia are overseen by the Therapeutic Goods Administration (TGA), which is part of the Australian Government Department of Health. The TGA is responsible for regulating therapeutic goods, including pharmaceutical drugs, medical devices, and other healthcare products, to ensure their safety, quality, and efficacy.
Pharmaceutical Regulations and Registration in Australia
Classification of drugs
Australia has a two-tiered system for the regulation of medicines such as Higher Risk and Lower Risk medicine. Within this regulatory framework, drugs are classified as Registered Medicine or Listed Medicine.
Registered Medicine – All drugs that are having a higher level of risk associated need to be registered with TGA. They must display an ‘AUST R’ number on the label as proof of registration and must be evaluated as either ‘high risk’ or ‘low risk’ registered. Registered medicine is further sub-classified as
- Prescription medicine (High risk) – For example sterile injectables
- Non-prescription medicine (Low risk) – Examples include mild analgesics, cough and cold medicines, and anti-fungal creams.
Listed Medicine – Listed medicines are typically considered mild, so sponsors are allowed to ‘self-assess’ their products in certain circumstances. Most of the listed medicines are self-selected by consumers and are used for self-treatment. All listed medicines
- Must display an ‘AUST L’ number on the label as proof of listing.
- Must NOT contain substances that are listed in the Poisons Standard.
- Must only contain permissible ingredients that are included in the Therapeutic Goods.
Pharma Clinical Trial Requirements
Key aspects of clinical trial requirements for drugs in Australia:
- Ethical Committee Approval: Before commencing a clinical trial, the research protocol must receive approval from the Human Research Ethics Committee (HREC). The HREC evaluates the trial’s design, potential risks, and benefits to participants, ensuring that the trial is conducted ethically and in accordance with the National Statement on Ethical Conduct in Human Research.
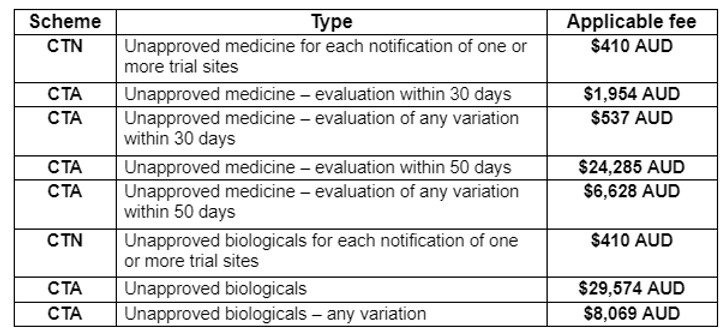
- Clinical Trial Notification (CTN) Scheme: The CTN scheme applies to most clinical trials of unapproved drugs and some early-phase trials of approved drugs. Under this scheme, sponsors are required to notify the TGA of their intent to conduct a clinical trial, and the trial can commence after 30 calendar days, provided there are no objections from the TGA or ethics committee. The CTN scheme may be chosen for earlier phase studies upon producing adequate safety information from preclinical studies.
- Clinical Trial Approval (CTA) Scheme: The CTA scheme applies to early-phase clinical trials of certain low-risk drugs and allows the trial to proceed without prior TGA approval. Instead, sponsors must notify the TGA, and the trial can commence after a waiting period. The CTA route is generally chosen for high-risk or novel treatments, such as gene therapy, where there is limited information on the safety aspects.
The sponsor is responsible for paying a fee to the TGA to submit an application for evaluation.
Good Clinical Practice (GCP): Clinical trials in Australia must adhere to the principles of Good Clinical Practice, which provide guidelines for the design, conduct, monitoring, recording, and reporting of trials. Compliance with GCP ensures data integrity and participant protection.
Informed Consent: Participants in clinical trials must provide informed consent before participating. The informed consent process includes providing detailed information about the trial’s purpose, procedures, potential risks and benefits, and the participant’s right to withdraw at any time.
Investigational Medicinal Product (IMP) Importation: If the drug being tested is not already approved in Australia, sponsors must seek approval from the TGA for the importation of the investigational medicinal product for use in the trial.
Monitoring and Reporting: Sponsors are responsible for monitoring the trial’s progress and ensuring that data is accurately collected and reported. Any adverse events or serious adverse events that occur during the trial must be reported to the TGA and ethics committee.
TGA Pharma Registration Process
The drug approval process in Australia does not mandate having legal representation. Drug sponsors have the option to engage legal counsel or regulatory consultants to assist and advise them throughout the registration process.
- Pre-submission Consultation: Before formally submitting an application, drug sponsors have the option to engage in a pre-submission consultation with the TGA. This consultation allows sponsors to seek advice and clarification on regulatory requirements, submission content, and any other aspects relevant to the drug application. It is an optional step.
- Compilation of Data: Drug sponsors must gather and compile comprehensive data on the drug’s quality, safety, and efficacy from preclinical studies and clinical trials. List of documents: Drug Registration Application Form; Chemistry, Manufacturing, and Controls (CMC) Data; Clinical Trial Data; Investigator’s Brochure (IB); GMP certificates; letter of authorization from the drug sponsor, details of any overseas approvals (if applicable); Certificate of Pharmaceutical Product (CPP)
- Submission of the Application: Once the data is compiled, the drug sponsor submits the application for drug registration to the TGA. The submission must include detailed information about the drug, its intended use, manufacturing processes, pharmacology, toxicology, clinical trial data, and proposed product labeling.
- Fees and Timelines: The registration process may take on an average of 330 calendar days or 11 months which includes the time for applicant activities.
For Prescription medicine – application and evaluation fee varies between $1241 AUD – $54,292 AUD & $4,954 – 217,598 AUD respectively.
For Listed medicine – application fee is approx. $1,300 AUD and evaluation fee varies from $16,299 AUD – $26,522 AUD depending on the type of ingredients in the drug.
- Evaluation by the TGA: The TGA conducts a thorough evaluation of the registration application to assess the safety, quality, and efficacy of the drug. This evaluation involves a review of all submitted data, including preclinical and clinical trial results.
- Advisory Committee Review (if applicable): For certain high-risk or novel drugs, the TGA may seek expert advice from an independent advisory committee, such as the Advisory Committee on Medicines (ACM). The committee provides additional expert input on the drug’s benefits and risks.
- Approval Decision: Based on the evaluation of the submitted data and, if applicable, the advice from the advisory committee, the TGA makes a decision on whether to approve the drug for registration. If approved, the drug will be included in the Australian Register of Therapeutic Goods (ARTG).
- Registration on the ARTG: Upon approval, the drug is registered on the ARTG, and the product is assigned an Australian Registration Number (Aust R) or an Australian Listed Medicine Number (Aust L) for prescription and over-the-counter drugs, respectively.
- Ongoing Post-Market Monitoring: After registration, the TGA continues to monitor the safety and quality of the drug through post-market surveillance and adverse event reporting.
Approved pharmaceuticals can be beneficial and may expedite the drug registration process in Australia to some extent. This is because regulatory authorities in different countries often rely on the evaluation and approval processes of reputable international regulatory agencies such as the USFDA and the EMA when considering drug applications in their own jurisdictions. Here’s how approval in the EU/US can help in progressing drug registration in Australia:
Reference to International Approvals: The Therapeutic Goods Administration (TGA) in Australia may accept certain assessments, evaluations, or approvals from stringent regulatory agencies like the European Medicines Agency (EMA) in the EU or the Food and Drug Administration (FDA) in the US as part of the evidence package submitted for drug registration in Australia.
Expedited Review Pathways: The TGA may offer expedited review pathways or fast-track options for drugs that have already been approved in other highly regulated markets like the EU or the US.
Utilization of Data: The clinical trial data and safety profiles that have been assessed and approved by EU or US regulatory agencies can be used as supportive data for the drug’s safety and efficacy during the Australian evaluation process.
International Collaboration: Regulatory authorities worldwide, including the TGA, often engage in international collaborations and harmonization efforts to exchange information and align regulatory processes. This can facilitate the acceptance of foreign approvals and reduce duplicative work.
Similarity Assessments: For generic drugs seeking registration in Australia, approval in the EU or US can support a similarity assessment, demonstrating bioequivalence to the reference product, which is often a prerequisite for generic drug approval.
Post Marketing Surveillance
Post-marketing surveillance, also known as pharmacovigilance, is a crucial aspect of drug regulation in Australia. It involves monitoring the safety of pharmaceutical drugs after they have been approved and are available in the market. The primary purpose of post-marketing surveillance is to identify and assess any previously unknown or rare adverse reactions to drugs and to take appropriate regulatory actions if necessary. The requirements for post-marketing surveillance of drugs in Australia include:
Adverse Event Reporting: Healthcare professionals, consumers, and drug sponsors are required to report any adverse reactions or side effects associated with pharmaceutical drugs to the Therapeutic Goods Administration (TGA) through the Adverse Medicine Events (AME) Line. The TGA encourages the reporting of both suspected and confirmed adverse events, regardless of the severity.
MedWatch Forms: The TGA provides MedWatch forms for reporting adverse events related to medicines. These forms facilitate the collection of essential information about the adverse event, the drug, and the patient.
Electronic Reporting: The TGA encourages electronic reporting of adverse events through various electronic systems to streamline the reporting process and enhance data collection and analysis.
Data Collection and Analysis: The TGA conducts audits to collect and analyse the reported adverse events to identify patterns, trends, and potential safety signals related to specific drugs or drug classes. TGA demands Corrective action and preventative actions (CAPA) for any potential risks or hazards.
Signal Detection: The TGA uses sophisticated methods to detect potential safety signals from the reported adverse events data. These signals trigger further investigations or actions as required.
Risk Management Plan (RMP) Review: For certain high-risk drugs, the TGA may require a risk management plan as part of the initial approval process. The RMP outlines strategies for ongoing safety monitoring and risk minimization during the post-marketing phase.
Risk Communication: The TGA communicates safety-related information, such as product recalls, safety alerts, or updates to product information, to healthcare professionals and the public when necessary.
Post-Market Reviews: The TGA may conduct periodic post-market reviews of specific drugs or drug classes to assess their safety, effectiveness, and ongoing benefit-risk profile.
Collaboration with International Agencies: The TGA collaborates with international regulatory agencies and participates in global pharmacovigilance efforts to share safety information and enhance surveillance.
References:
- https://www.tga.gov.au/medicines-and-tga-classifications
- https://www.tga.gov.au/sites/default/files/australian-clinical-trial-handbook.pdf
- https://clinregs.niaid.nih.gov/country/australia#scope_of_assessment
- https://www.pharmaceutical-technology.com/sponsored/regulatory-requirements-for-clinical-trials-a-comparison-of-australia-and-the-us/#:~:text=To%20conduct%20a%20clinical%20trial,Clinical%20Trial%20Exemption%20(CTX).
- https://www.statista.com/outlook/hmo/pharmaceuticals/australia#:~:text=The%20projected%20revenue%20in%20the,US%241.80bn%20in%202023.

